Article Text

Abstract
Phaeochromocytoma is a rare clinical entity in children. Contrary to traditional teaching, which suggested that 10% of phaeochromocytomas are “familial”, a germline mutation has been identified in up to 59% (27/48) of apparently sporadic phaeochromocytomas presenting at 18 years or younger and in 70% of those presenting before 10 years of age. The inherited predisposition may be attributable to a germline mutation in the Von Hippel–Lindau gene, the genes encoding the subunits B and D of succinate dehydrogenase, the RET proto-oncogene predisposing to multiple endocrine neoplasia type 2, or the neurofibromatosis type 1 gene. Of these, the Von Hippel–Lindau gene is the most commonly mutated gene in children presenting with a phaeochromocytoma. Genetic counselling is recommended before gene testing and investigation of the wider family. This review provides guidance on the aetiology, investigation, management, histopathology, genetics and follow-up of children with a phaeochromocytoma.
Statistics from Altmetric.com
Phaeochromocytoma is a rare clinical entity in children. Contrary to traditional teaching, which suggested that 10% of phaeochromocytomas are “familial”, advances in molecular genetics have revealed an identifiable germline mutation in up to 59% (27/48) of apparently sporadic phaeochromocytomas presenting at 18 years or younger and in 70% of those presenting before 10 years of age.1
The inherited predisposition may be attributable to a germline mutation in the Von Hippel–Lindau (VHL) gene, the genes encoding the subunits B and D of succinate dehydrogenase (SDHB and SDHD), the RET proto-oncogene predisposing to multiple endocrine neoplasia type 2 (MEN2) or the neurofibromatosis type 1 (NF1) gene. Of these, the VHL gene is the most commonly mutated gene in children presenting with a phaeochromocytoma.1 2 Referral to the clinical genetics department is recommended for genetic counselling before gene testing and investigation of the wider family. This review provides guidance on the aetiology, investigation, management, histopathology, genetics and follow-up of children with a phaeochromocytoma.
Phaeochromocytomas are tumours arising from the catecholamine-producing chromaffin cells in the adrenal medulla. Tumours arising in extra-adrenal sympathetic and parasympathetic paraganglia are closely related and are classified as extra-adrenal paragangliomas.3 Phaeochromocytomas and extra-adrenal sympathetic paragangliomas usually secrete catecholamines, whereas parasympathetic paragangliomas of the head and neck are usually non-functioning (table 1). For the purpose of this review, secretory extra-adrenal paraganglioma are included under the term phaeochromocytoma.
Data on the aetiology, diagnosis and management of phaeochromocytoma in children are limited. Guidance is based on observational studies and consensus opinion.
EPIDEMIOLOGY
Phaeochromocytomas are thought to be responsible for ∼1% of childhood hypertension.4 The National Registry of Childhood Cancers (1981–2002) suggests a minimum estimated incidence of benign and unspecified phaeochromocytoma of 0.11 per million children.5
The incidence of malignant phaeochromocytoma is approximately 0.02 per million children.5 Reports from three studies of 24, 50 and 58 paediatric patients suggest a varying malignancy rate of 25%, 56% and 12%, respectively.2 6 7 The same three studies report bilateral phaeochromocytoma in 25%, 32% and 34% and extra-adrenal functional catecholamine-secreting tumours in 8%, 18% and 22%, respectively. The presence of malignancy and extra-adrenal or bilateral disease depends on the underlying genetic disorder if present. Difficulties in confirming the presence of malignancy as opposed to multiple primary tumours may also contribute to the disparity between the three cohorts.
Data on recurrence risks are also lacking for these reasons. One study suggested a 12% recurrence rate for “true phaeochromocytoma” in children, although the authors concede that this may have been due to inadequate primary clearance.7
Phaeochromocytomas may present in the neonatal period,8 but are more common in older children.2 Data extrapolated from the National Registry of Childhood Tumours suggest a male preponderance in childhood, which is corroborated by other studies.2 5 9 There may be a female preponderance during the reproductive years, suggesting a hormonal influence, resulting in an equal sex incidence overall.2
PRESENTATION AND COMPLICATIONS
Phaeochromocytoma may present in a variety of ways ranging from vague symptoms to a hypertensive emergency. Symptoms may include headache, sweating, flushing, palpitations, blurred vision, syncope, panic attacks, tremor, gastrointestinal disturbance and weight loss. Pham et al10 report that 30% present with symptoms of mass effect or as an incidental finding on imaging. Some phaeochromocytomas may remain undiagnosed before death.11 Hypertension is the most consistent sign, which is sustained and without paroxysms in 63%.2
Complications include cardiomyopathy, hypertensive crisis, cerebrovascular accidents, convulsions, mass effect and multiorgan failure. Mortality from phaeochromocytoma depends on the presentation and presence of malignancy. Children with a phaeochromocytoma should be referred to a paediatric endocrinologist and a children’s cancer and leukaemia centre with appropriate medical and surgical expertise.5
BIOCHEMICAL DIAGNOSIS
Confirmation of the diagnosis requires the demonstration of inappropriate production of catecholamines (norepinephrine and epinephrine). This has traditionally been achieved by measuring 24 h urinary excretion of total metanephrines and catecholamines.12 13 The plasma concentrations of norepinephrine and epinephrine are determined by the release of these substances from the adrenal gland dependent on sympathoadrenal tone, whereas the concentrations of plasma metanephrines, their metabolites, are due to the metabolism of epinephrine and norepinephrine within the chromaffin cells and subsequent continuous release, which is far less dependent on sympathoadrenal tone. The two processes are independent of each other.14 15 Measurement of fractionated metanephrines (normetanephrine and metanephrine measured separately) in urine or plasma provides better diagnostic sensitivity than measurement of the parent catecholamines.16
Measurement of 24 h urinary total metanephrines and catecholamines (sensitivity 90%; specificity 98%) is appropriate when the patient is at low risk of phaeochromocytoma (isolated hypertension), whereas measurement of fractionated plasma metanephrines (sensitivity 97%; specificity 85%) may be the test of choice when the patient is at greater risk (genetic predisposition or adrenal mass).17 Lenders et al16 have suggested that demonstration of normal plasma fractionated metanephrines can virtually exclude a diagnosis of a phaeochromocytoma.
The recommendations of the British Society of Paediatric Endocrinology and Diabetes state that phaeochromocytoma should be confirmed by showing raised 24 h urinary metanephrines and catecholamines on two occasions.5 Tandem mass spectrometry for the measurement of metanephrines may become more readily available in the future and promises to be a more precise and relatively low cost method than the more readily available techniques of today.18
IMAGING PHAEOCHROMOCYTOMA
Successful management of phaeochromocytoma depends on high-quality imaging to accurately localise the tumour and stage the extent of disease.19 The choice of imaging modality depends on the level of suspicion determined by biochemical evidence, previous history of a phaeochromocytoma, and inherited predisposition. Ultrasound is a useful initial investigation in those who are symptomatic. However, an abdominal MRI or CT scan and a whole-body metaiodobenzylguanidine (MIBG) scan (123I) are advised for confirmation, accurate staging and preoperative planning.5 Unfortunately no single investigation can reliably predict malignancy before surgery. Routine selective venous sampling for localisation and stimulation tests are not indicated, neither is image-guided or open biopsy.5
Cross-sectional imaging can be used to localise the tumour more accurately in three planes. CT is fast and sensitive, but involves ionising radiation. A standard CT scan of the abdomen would normally be enhanced with intravenous iodinated contrast. Iodinated contrast is related to hypertensive crises in patients with phaeochromocytoma, so if there is suspicion of phaeochromocytoma before imaging, contrast should be avoided and an unenhanced scan performed. MRI is a more lengthy investigation and may require sedation or a general anaesthetic. However, phaeochromocytoma has a more typical unenhanced appearance on MRI than on CT and therefore the former may be the investigation of choice.
MRI and CT are good modalities for identifying a lesion. In a study of 236 adult patients, these techniques failed to detect 0% and 5.8%, respectively, of benign paraganglioma.20 However, they lack specificity. Functional imaging with 123I-labelled MIBG scintigraphy may lack sensitivity (80–90%), but has high specificity (98%).18 21 Abnormal neuroectodermal tissue takes up the isotope and gives a focal area of increased uptake on the scan (fig 1). This is useful for confirming the presence of chromaffin tissue and aids in locating extra-adrenal paragangliomas or multiple synchronous primaries.

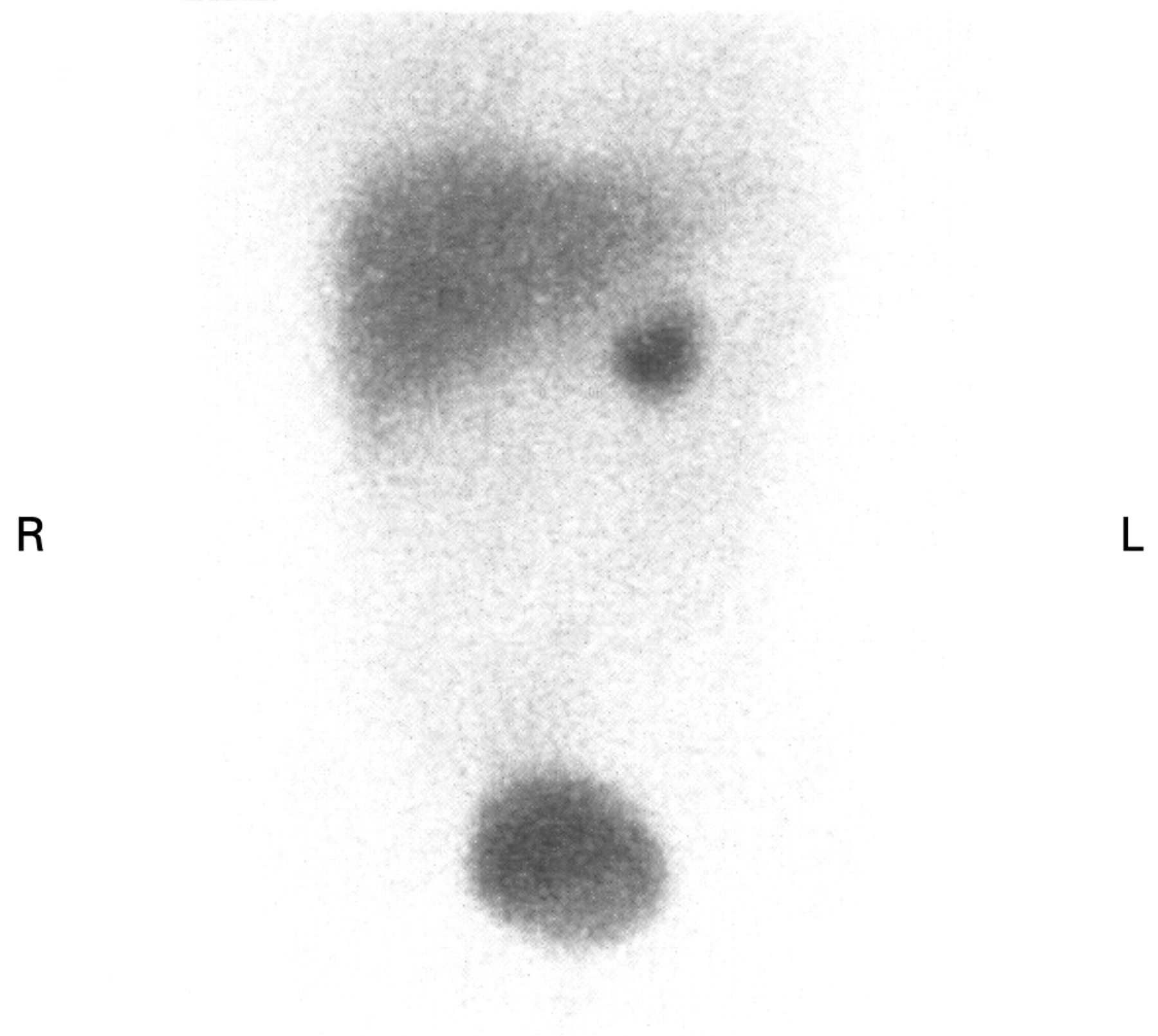{kind=link}
Functional [18F]fluoro-2-deoxyglucose/6-[18F]fluorodopamine positron emission tomography has produced promising results, suggesting that multimodality imaging is appropriate in tumours that are difficult to localise.22 23 MIBG studies with correlative single photon emission CT/CT fusion imaging are beneficial in cases where the diagnosis is equivocal and can increase the diagnostic certainty by up to 90%.24 However, the increased radiation exposure should be considered.25
GENETICS
Traditional teaching suggested that 10% of phaeochromocytomas are “familial”.5
Recent advances in molecular genetics, however, have revealed an identifiable germline mutation in up to 59% (27/48) of apparently sporadic phaeochromocytomas presenting at 18 years or younger and in 70% of those presenting before 10 years of age.1
As discussed above, the inherited predisposition may be attributable to a germline gene alteration in the VHL, SDHB, SDHD, MEN2 or NF1 genes (table 2). Further advances in molecular genetics are likely to identify additional predisposition genes. For example, mutations in the subunit C of succinate dehydrogenase (SDHC) are usually associated with non-secretory, parasympathetic paraganglioma and are not routinely screened for. However, Mannelli et al26 described a 15-year-old patient with a norepinephrine-secreting abdominal paraganglioma with a novel, nonsense SDHC mutation.
Box 1 Suggested preoperative medical blockade of phaeochromocytoma
2 weeks before surgery
-
α-Adrenergic blockade
-
-
Oral phenoxybenzamine
-
Age 1 month–18 years
-
0.5–1 mg/kg twice daily adjusted according to response
-
-
β-Adrenergic blockers
-
-
Standard dosing regimen
-
Given to patients after adequate α blockade
-
Administration of β blocker before α blockade can worsen hypertension secondary to unopposed vasoconstriction
-
Metyrosine
-
-
Not licensed in UK
-
Not tested in children under 12 years of age; available on named patient basis
-
Dose 250 mg every 6 h to a maximum of 4 g a day orally
-
May not be necessary for patients with minimal or no symptoms from a minimally functioning phaeochromocytoma
-
Night before surgery
-
Intravenous fluids are necessary to maintain adequate hydration and blood volume
-
At midnight give phenoxybenzamine at 1 mg/kg and, where indicated, metyrosine 1 g orally
Adapted from Walter et al.31
VHL is the most commonly mutated gene predisposing to a phaeochromocytoma or extra-adrenal paraganglioma in children, followed by SDHB, SDHD, RET and NF1.1 5 However, this may be liable to some geographical variation. Testing is usually conducted in this order unless there are other features or a family history to suggest otherwise. NF1 should be clinically detectable, and molecular investigations are rarely necessary.
Gene alterations in RET, NF1, VHL or SDHB/D may predispose to phaeochromocytoma or paraganglioma by changing the normal developmental apoptotic pathway of sympathetic neuronal precursor cells.27 They are all inherited in an autosomal dominant manner. If a person has the gene alteration, there is a 50% chance of transmitting it to each of their children. Often there is no obvious family history at presentation because the gene alteration has occurred in the affected person for the first time, the gene alteration exhibits reduced penetrance, or the family history has not yet been ascertained.
The risk of developing a phaeochromocytoma depends on the underlying condition (table 2). In one study, about one-third of patients of all ages found to carry a germline mutation developed a lesion typical of the mutation during follow-up or had a member of the family identified with a lesion after screening.1
Blood can be taken at the time of surgery for DNA storage (5–10 ml in an EDTA bottle). However, children and their families should be referred to the clinical genetics department for assessment, including a three-generation family history, clinical examination for NF1 and genetic counselling, before molecular analysis for the VHL, SDHB/D and RET gene alterations. In situations where mutation analysis is not readily available, further investigation of the index case and first-degree relatives may be required to clinically reduce the risk of an underlying genetic condition and direct screening.
PREOPERATIVE MANAGEMENT
Phaeochromocytoma may present as an emergency. Management is symptom-dependent usually requiring pharmacological intervention to block the effects of high concentrations of circulating catecholamines. Hypertensive crisis is the most common emergency associated with phaeochromocytoma and may be defined as “high blood pressure resulting in life threatening emergencies or compromises of vital organ function”.28 Blood pressure should be controlled at a rate sufficient to reduce toxicity, but avoiding acute hypoperfusion of vital organs. This should be undertaken in a setting where invasive monitoring is available to assess the central nervous system, cardiac and renal function.29 Intravenous agents are preferable under these circumstances, to allow adjustment of infusion rate according to response. A continuous infusion of phentolamine or sodium nitroprusside is preferable.30 Intravenous labetalol by bolus injection, followed by continuous infusion, may also be used.29 In patients with hypertension but without signs of end-organ damage, oral anti-hypertensive drugs can be considered.
Surgery is the curative option for most patients. Preoperative preparation is important in order to minimise the complications arising from surgery and may take several weeks. Induction of anaesthesia and manipulation of the tumour causes unpredictable release of catecholamines, which may result in hypertensive crisis, stroke and arrhythmias. To prevent these problems, patients must undergo pharmacological blockade of catecholamine synthesis or effects before surgery.31
Routine preoperative α-adrenoceptor blockade opposes catecholamine-induced vasoconstriction and its sequelae.28 A β-adrenoceptor blocker is added to oppose the reflex tachycardia often associated with α blockade. β blockade alone can be dangerous and is contraindicated, because it does not prevent, and can actually augment, effects of catecholamines at α-adrenoceptors, resulting in hypertensive crisis.19 30 32 It is important that, if β-adrenoceptor blockers are used, they should be used only after adequate pretreatment with α-adrenoceptor antagonists. Adequate α blockade is indicated by normotension or the development of side effects such as orthostatic hypotension, tachycardia, nasal congestion, nausea and abdominal pain.28 A retrospective review of α blockade with phenoxybenzamine, prazosin or doxazosin in preoperative preparation concluded that surgery is safe with each of these drugs.33 α-Methyl-para-tyrosine (metyrosine) competitively inhibits tyrosine hydroxylase, the rate-limiting step in catecholamine biosynthesis.34 Treatment with metyrosine reduces tumour stores of catecholamines, decreases the need for intraoperative antihypertensive drugs, lowers intraoperative fluid requirements, and attenuates blood loss.35
A combination of metyrosine, phenoxybenzamine, β blocker and liberal salt intake starting 10–14 days before surgery leads to better control of blood pressure and decreases surgical risks (box 1). Combined medical blockade also allows relaxation of the constricted vascular tree and expansion of the reduced plasma volume, thus avoiding hypotension after sudden diffuse vasodilation at the time of tumour removal. Intravenous fluids 24 h before surgery may be necessary to hydrate and ensure adequate blood volume.32
Alternative agents and approaches have been used successfully in limited settings. The oral calcium channel blocker, nicardipine (3–10 days before surgery), has been used to control intraoperative blood pressure, but it did not prevent all haemodynamic instability during tumour resection.36 Angiotensin-converting enzyme inhibitors have also been used successfully to manage hypertension in older patients with phaeochromocytoma.37
ANAESTHETIC MANAGEMENT
Meticulous anaesthesia throughout the operative procedure is crucial to reduce the risk of hypertensive crisis during tumour manipulation and severe hypotension after removal. Pre-medication may be necessary to ensure that the patient is calm in the anaesthetic room. Invasive monitoring of BP should be started before or (more realistically in this population) immediately after induction of anaesthesia. A combined general anaesthetic and epidural technique is useful both during the operation (to reduce the effects of vasoactive substances released during tumour handling) and for postoperative analgesia. Acute episodes of hypertension during surgery can be controlled with sodium nitroprusside by infusion. Hypotension may occur when venous drainage of the tumour is removed; infusions of dobutamine and/or norepinephrine may be required to maintain normotension for a variable period after surgery (typically reducing after the first 2 h).
SURGICAL MANAGEMENT
Traditionally, open surgery for an adrenal or abdominal/pelvic (organ of Zuckerkandl) primary tumour (95% of cases) permits full inspection of the lesion and, in cases of suspected malignancy, the opportunity to assess loco-regional disease, vascular element invasion and metastases. Thoracic tumours can be approached via a muscle-sparing thoracotomy. Those located in rarer sites include the head/neck, urinary bladder or central nervous system. The latter will require neurosurgical expertise. Advantages of open operation are that complete tumour resection is facilitated with early isolation and control of venous drainage, thereby minimising systemic catecholamine release.38
Paravertebral sympathetic chain paragangliomas and retrocaval extra-adrenal tumours can be particularly challenging, with careful dissection and mobilisation of great vessels required to expose the “hidden” offending lesion. Vascular reconstruction may be needed in these technically demanding scenarios. Ex vivo techniques (“bench surgery”) have recently been described by transplant surgeons to achieve resection of tumours located in close proximity to the portal triad anatomy and inferior vena cava.39
Total adrenalectomy for bilateral tumours carries substantial morbidity, with long-term requirement for steroid hormone replacement therapy, risks of osteoporosis, and Addisonian crises if patient compliance lapses.40 Adrenal “cortical sparing” procedures have been advocated for patients with bilateral tumours.41
Advances in minimally invasive surgery have increased the feasibility and safety of laparoscopic adrenalectomy for non-malignant tumours (<10 cm), with expanding indications for a laparoscopic approach in childhood.41 42 This strategy has also been used recently to resect extra-adrenal tumours.43
HISTOPATHOLOGY
Histopathological examination of the tumour is essential to confirm the diagnosis and site of origin and to assess features that may indicate a greater likelihood of malignancy or syndromic disease (table 2).44 45
Phaeochromocytomas and extra-adrenal paragangliomas can have identical histological appearances. Microscopic evaluation of peri-adrenal tumours is therefore important to confirm extra-adrenal origin, which is associated with an increased risk of malignancy.46 Differentiation from other tumour types is usually straightforward with the use of immunohistochemistry.47 Occasionally, composite tumours with areas of ganglioneuroma or ganglioneuroblastoma are identified.46
The biological behaviour of these tumours cannot be accurately predicted on the basis of histopathological features. Malignancy can only be diagnosed with documented metastatic disease in a site where chromaffin cells are usually absent, to exclude the possibility of a synchronous or metachronous primary tumour.3 Recent systematic attempts to quantify risk of malignancy on the basis of histopathological features have included paediatric patients in both benign and malignant groups.46–49 These studies use a combination of factors, such as growth pattern, necrosis, mitotic rate and tumour cellularity, to predict the likelihood of malignant behaviour. Although none are perfectly predictive, they represent the best evidence currently available for stratifying risk, and some features are incorporated into the reporting guidelines of the Royal College of Pathologists and the Association of Directors of Anatomic and Surgical Pathology in the USA.50 51
Tumour expression of many other proteins, including tenascin, cyclo-oxygenase-2 and vascular endothelial growth factor, has been found to correlate with malignant behaviour.52–54 There is a greatly increased risk of malignancy in the context of SDHB mutation, and extra-adrenal location is increased in SDHD and SDHB mutations and in VHL syndrome.44 55 56
MANAGEMENT OF MALIGNANT PHAEOCHROMOCYTOMA
Surgical excision remains the treatment of choice for malignant disease, but medical treatments have been explored in adults using 131I-labelled MIBG57 and somatostatin analogues58 and combination chemotherapy.59 131I-labelled MIBG is the most promising of these, leading to an increase in survival for those responding in the first 6 months of treatment. It has been suggested that that myeloablative chemotherapy with stem cell rescue may have a place in the treatment for malignant disease, but it is yet to be fully evaluated. Multicentre studies are therefore crucially needed to achieve international consensus on dose schedules versus other combination therapies.60
FOLLOW-UP
Follow-up is life long to detect the possible development of metachronous tumours and tumours at other sites. It is partly determined by the underlying genetic condition if present (table 2). Screening schedules for the different genetic disorders remain controversial and should be overseen by a paediatric endocrinologist or person with the relevant expertise. Initial follow-up should include 6-monthly measurement of blood pressure and urinary catecholamines. Annual follow-up may be appropriate later5 and for children in whom a gene alteration is not identified; whether they require further imaging and its frequency are contentious issues. Children who have undergone bilateral adrenalectomy require regular assessment to monitor adequacy of steroid replacement.
If a second or recurrent phaeochromocytoma is identified, investigation is as for the initial presentation. If a mutation was not identified initially, a clinical geneticist should review the child again.5 If a gene alteration is identified, family members should be offered a referral to the clinical genetics department.
Liaison between the paediatric and adult sectors is vital to ensure continued screening. Data collection through central registration is important to increase understanding of phaeochromocytoma and predisposing conditions, thus facilitating effective screening and management.5
REFERENCES
Footnotes
-
Competing interests: None.