Article Text

Abstract
Proteomics is the study of a large number of proteins in biological systems. We aim to introduce the complex field to paediatricians and present some recent examples of applications to paediatric problems. Various approaches have been used to study proteomes. The current mainstay is tandem mass spectrometry of enzymatically digested proteins (‘bottom-up proteomics’), and we describe the experimental and computational approach further. Proteomics can offer advantages over transcriptomics by giving direct information about proteins rather than RNA; however, typically data are obtained at lower depth and the confident identification of mass spectra can be challenging. Proteomics frequently complements transcriptomics and other -omics. Used effectively, proteomics offers promise to help answer important clinical and biological questions.
- proteomics
- paediatrics
Data availability statement
There are no data in this work
Statistics from Altmetric.com
The term ‘proteome’ was coined in a 1995 study of bacterial proteins.1 In accordance with the common approach of -omics sciences, proteomics is the study of a large number of proteins in biological systems (eg, cells, tissues or organisms) at once.
The simplicity of this statement belies the complexity of the field, since an enormous range of basic questions can be asked regarding proteins: about their abundance, localisation, interactions, modification states and folding, through time, under different conditions and in different subjects. Regardless, to date, the majority of clinical proteomic studies focus on answering biological questions based on protein abundance and post-translational modifications.
Techniques
Early proteomic studies relied on separating complex protein mixtures on 2D gels, and this approach still has a role.2 Individual proteins can be resolved as spots and identified by time-consuming sequencing by Edman degradation or mass spectrometry (MS). Abundance changes can be recognised through changes in spot intensity.
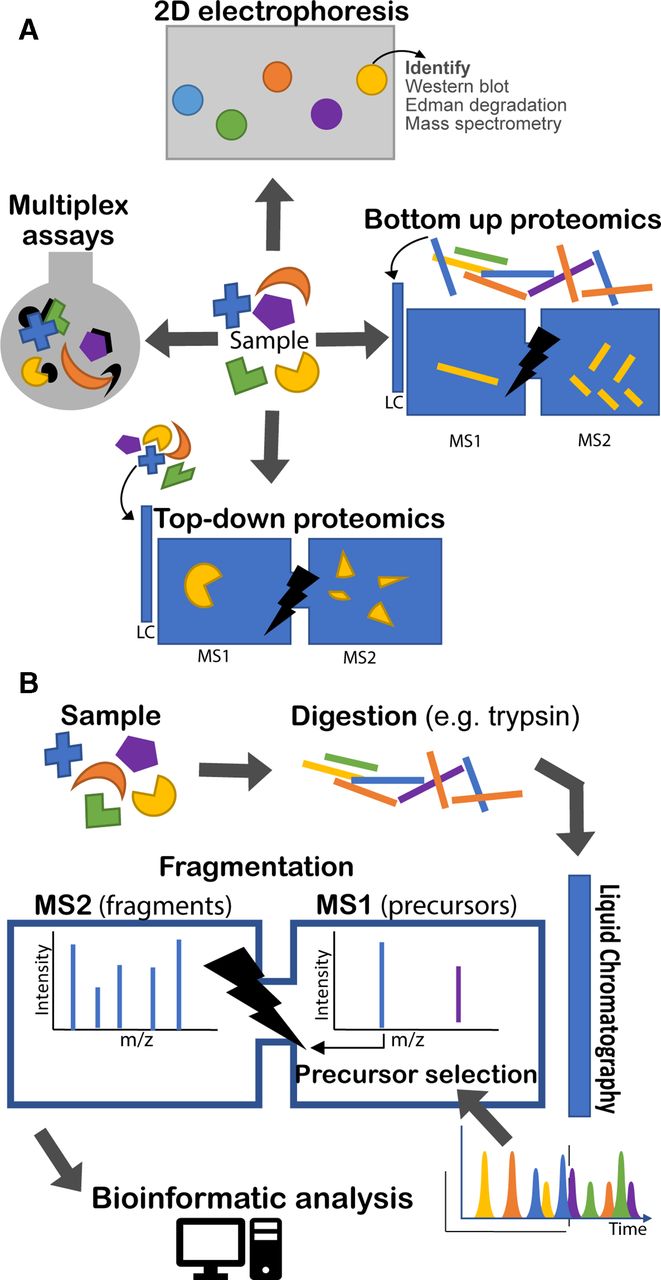
Development of tandem MS and allied informatics allowed unfractionated protein mixtures to be analysed in one run. Presently tandem MS of digested proteins (‘bottom-up proteomics’) is the main approach,3 though others are used (figure 1A).
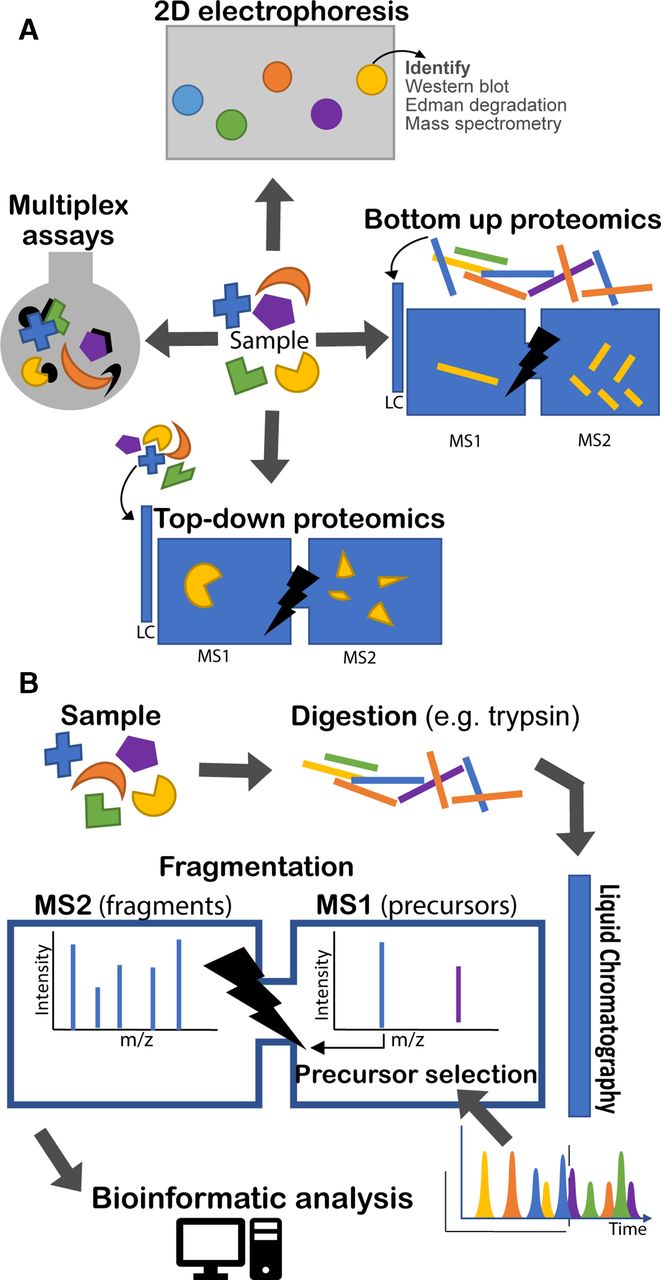
Proteomic techniques illustrated. (a) Four types of approach are illustrated. 2D electrophoresis separates proteins on a gel by electrochemical means. With subsequent staining, protein ‘spots’ can be recognised and compared between samples. Identification of proteins requires other tools, such as Western blotting or MS. Top-down and bottom-up proteomics both rely on tandem (multiple rounds of) MS. The bottom-up approach analyses peptides produced by enzymatic digest of proteins. The top-down approach analyses intact proteins directly. In both cases a second round of MS is undertaken following fragmentation (MS2). Finally, various multiplex assay technologies exist for quantifying panels of proteins in solution simultaneously (eg, Luminex and Mesoscale Discovery). (b) The bottom-up proteomic approach is illustrated in more detail. A sample is digested, usually by trypsin. The various peptides in the sample are eluted over time through a liquid chromatography column and ionised into the mass spectrometer. The mass spectrometer regularly analyses the eluted peptide species (MS1) and selects a number of precursors to be sequentially fragmented and analysed (MS2). Bioinformatic analysis is required to identify peptides from their precursor and fragment mass spectra (MS1 and MS2). LC, liquid chromatography, MS, mass spectrometry.
Mass spectrometry-based proteomics
Figure 1B illustrates the process of bottom-up proteomics which is described in more detail by Zhang et al.3 Proteins are extracted from a sample and digested (most often with trypsin). The digested peptides are separated by liquid chromatography and eluted over time and directly introduced into the mass spectrometer detector through the ionisation interface. Successive rounds of peptide ions (‘precursors’) are subjected to a first round of MS (usually at high resolution) to measure their mass-charge ratio (m/z). Typically, the most abundant ions are then selected to be fragmented in turn and undergo a further round of MS.
Various bioinformatic approaches exist to identify peptides and proteins from the data.4 Most commonly, the fragment mass spectra are matched to possible peptides from a relevant protein database, controlling for chance matches. Proteins can be quantified by inspecting the intensity of their corresponding precursor signals.
There is increasing interest in top-down proteomics, in which complete proteins are analysed by tandem MS, with an intervening fragmentation step. There are advantages for detection of protein isoforms. However, there are challenges in dealing with complex protein mixtures, and it is most frequently applied to individual proteins. Interested readers are directed to a recent review.5
The value and challenge of proteomics
One may ask what value proteomics can add when the field of transcriptomics is very advanced. The great strength of proteomics is that it provides direct information about the structural, signalling and enzymatic building blocks of the body, and proteins comprise a majority of drug targets. In contrast, transcriptomics tells us about abundance of individual transcripts, but not directly about protein abundance or state. The correspondence between gene and protein expression levels is often poor; the two techniques provide complementary information and are frequently used together.6
Protein-based diagnostic tests are potentially more easily translatable into low-cost near-patient technologies. Such tests are already in wide use, for example, pregnancy tests and rapid-diagnostic tests for malaria.
However, the confident and comprehensive identification and quantification of peptides and then proteins from mass spectra is not without challenge. Unlike sequencing technologies which directly read nucleotides with confidence scores, further steps are necessary to assign fragment mass spectra to putative peptides and proteins. Further, where typical sequencing runs give tens of millions of reads per sample, typical proteomic studies obtain only tens of thousands of fragment spectra per sample.
Applications
Diagnostic and prognostic markers
Diagnosis of sepsis and necrotising enterocolitis (NEC) on the neonatal unit remains challenging clinical problems. MS-based proteomic analysis of neonates’ urine yielded seven proteins which were then validated in a separate cohort by ELISA as protein biomarkers for diagnosis of sepsis, and prognosis and diagnosis of NEC.7 Areas under the curve in a validation cohort indicated good performance (all >0.95), with sensitivity and specificity for NEC versus sepsis of 89% and 80%, respectively.
There is increasing drive to target treatments to individual patients by deeper phenotyping (so-called ‘personalised’ or ‘precision’ medicine), especially in oncology. Jiang et al 8 identified a large number of proteins differentially expressed between prednisolone-resistant and prednisolone-sensitive acute lymphoblastic leukaemia cell lines. One of these, PCNA, was validated as a biomarker of prednisolone responsiveness in children, independent of subtype.
Understanding biology and disease
It is believed that cow’s milk antigens enter maternal milk to drive cow’s milk protein allergy in exclusively breastfed infants. However, antibody-based techniques can miss digested proteins and cross-reactivity cannot be excluded. Zhu et al 9 recently identified 36 non-human proteins in human breastmilk, mostly of bovine origin, using bottom-up proteomics. A further study10 showed that cow’s milk peptides in breastmilk peak at 2 hours postingestion and that detected peptides are rich in proline which may have helped them resist digestion in the gut.
The factors which determine severity of malaria remain poorly understood. Reutersward et al 11 used an antibody bead array approach to characterise the serum proteome of children with malaria and healthy controls. Thirty-seven proteins were found to be differentially abundant in severe and non-severe malaria; these included acute phase reactants and proteins involved in cell migration/adhesion and tissue remodelling.
Closing remarks
In this brief review, we have introduced proteomic principles and technologies, and a few examples of their application.
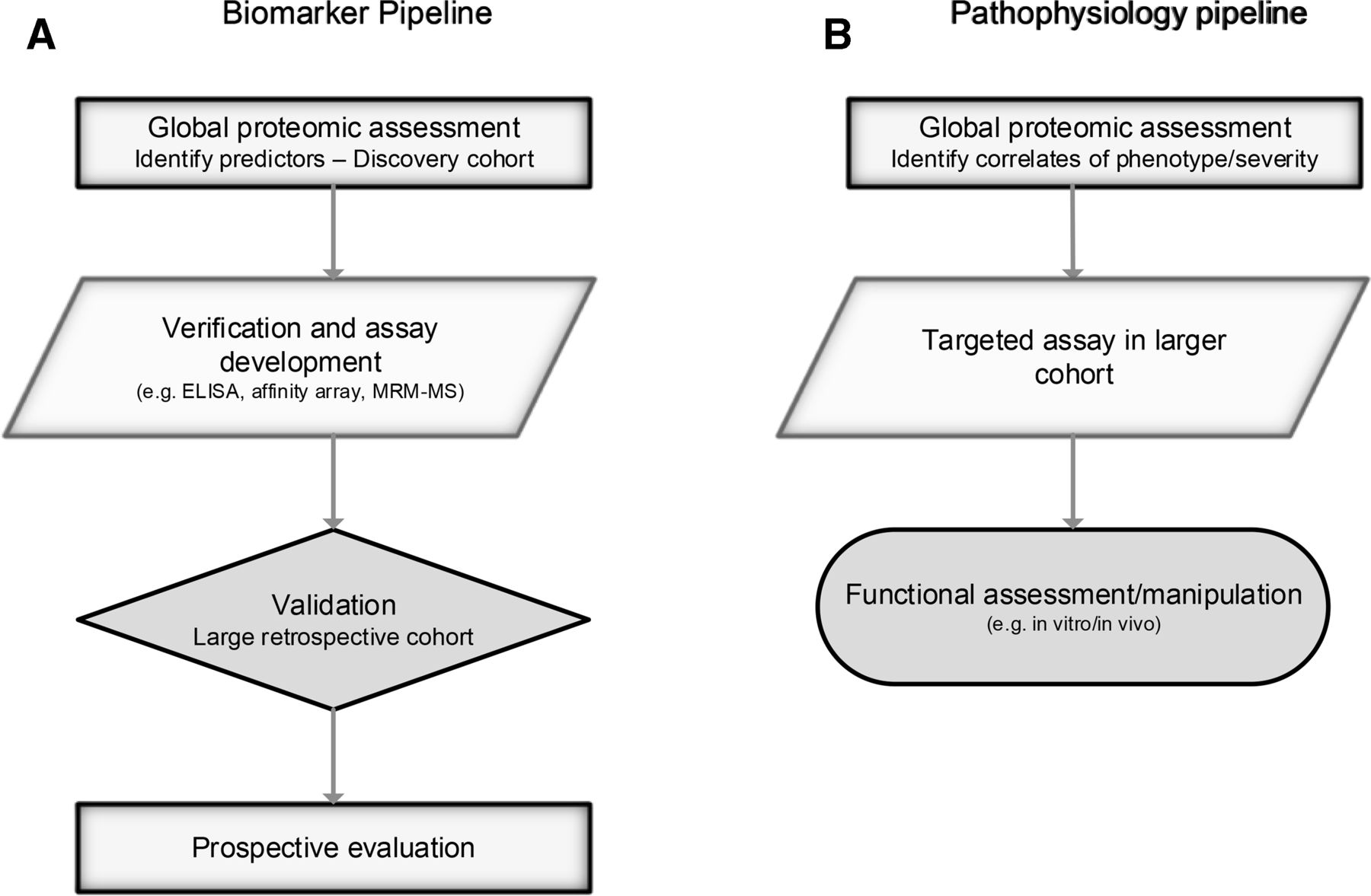
In common with all -omics, the rapidity of progress and application can be overstated, the volume of data leads to computational and statistical challenges, and techniques can be employed thoughtlessly or simply because they are in vogue.12 Identifying biomarkers is not enough—the road to clinical application is long, as outlined in recent reviews and shown in figure 2.13 14 Helpfully, data sharing is increasingly mandated, allowing validation, reanalysis, repurposing and meta-analysis (eg, PRIDE archive).15
{kind=link}
{kind=link}
Proteomics can be used to identify biomarkers and understand pathophysiology. Multiple steps are required to ensure validity, and for biomarkers, clinical utility. Outline processes are illustrated. Further detail on biomarker discovery is given by Parker and Borchers (2014).
Used effectively (and often complementing other ‘-omics’ techniques), proteomics offers promise to help answer important clinical and biological questions.
Data availability statement
There are no data in this work
References
Footnotes
Contributors AJM wrote the draft of the manuscript. AJM and SM finalised the manuscript. SM prepared figures.
Funding AJM is funded through a Wellcome Trust/Imperial College London 4i PhD Fellowship. SM is funded through a Biomedical Research Centre Institute for Translational Medicine and Therapeutics grant.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.