Article Text

Abstract
There have been phenomenal advances in our understanding of renal biology over the last 20 years through our ability to define the genetic mutations causing kidney disease in children. This review will take you through a trip down the nephron and highlight how these conditions may present to the paediatrician and the molecular basis for their biological effects.
- Basic Science
- Nephrology
Statistics from Altmetric.com
Introduction
This article travels through the nephron to outline the clinical features in children with genetic kidney diseases in the context of applied physiology. These inherited conditions commonly first present to the paediatrician and we discuss how they present and cover some of the well-known and recently discovered genetic mechanisms of disease.
Identifying the genetic causes of disease
The way we are identifying disease causing genetic mutations has changed over recent decades and continues to evolve as technology and availability improves. Previously, the genes responsible for Mendelian diseases were identified using linkage analysis with large extended families which commonly had a number of generations affected with the phenotype in question. This process was a lengthy one as a genome-wide search is required before the genetic locus responsible is found, followed by time consuming sequencing the region in cases and controls to find the mutations causative for disease.1 Indeed it could take several years to identify a disease causing mutation in an informative family.
Over the past decade the major advance in genetic technology had been the introduction of next-generation sequencing (NGS). This technology is now enabling us to test blood samples quickly and affordably to determine some genetic causes of disease. An example of this in paediatric nephrology is the use of NGS in determining whether a genetic mutation is present in a child who has steroid resistant nephrotic syndrome.2 This information has an impact on treatment and prognosis and is just one of the potential clinical applications of NGS. See the review (What is next generation sequencing?) in Education and Practice for further information about advances in genetic technology.2a
The nephron
Figure 1 shows the nephron and the sections affected by genetic diseases mentioned in this article. The nephron is the physiological functional unit of the kidney and starts with the glomerulus where ultrafiltration occurs; many of the diseases affecting the glomerulus are related to the breakdown of this filtration barrier. The tubules are the next component of the nephron, composed of the proximal tubule, loop of Henle and distal tubule. These are responsible for water and electrolyte homeostasis, as well as the re-absorption of other plasma components. Tubular disease is therefore often related to electrolyte disturbance or failure of re-absorption in the tubules. The final part of the nephron is the collecting duct, where concentration of the urine occurs before it enters the renal pelvis and the lower urinary tract.
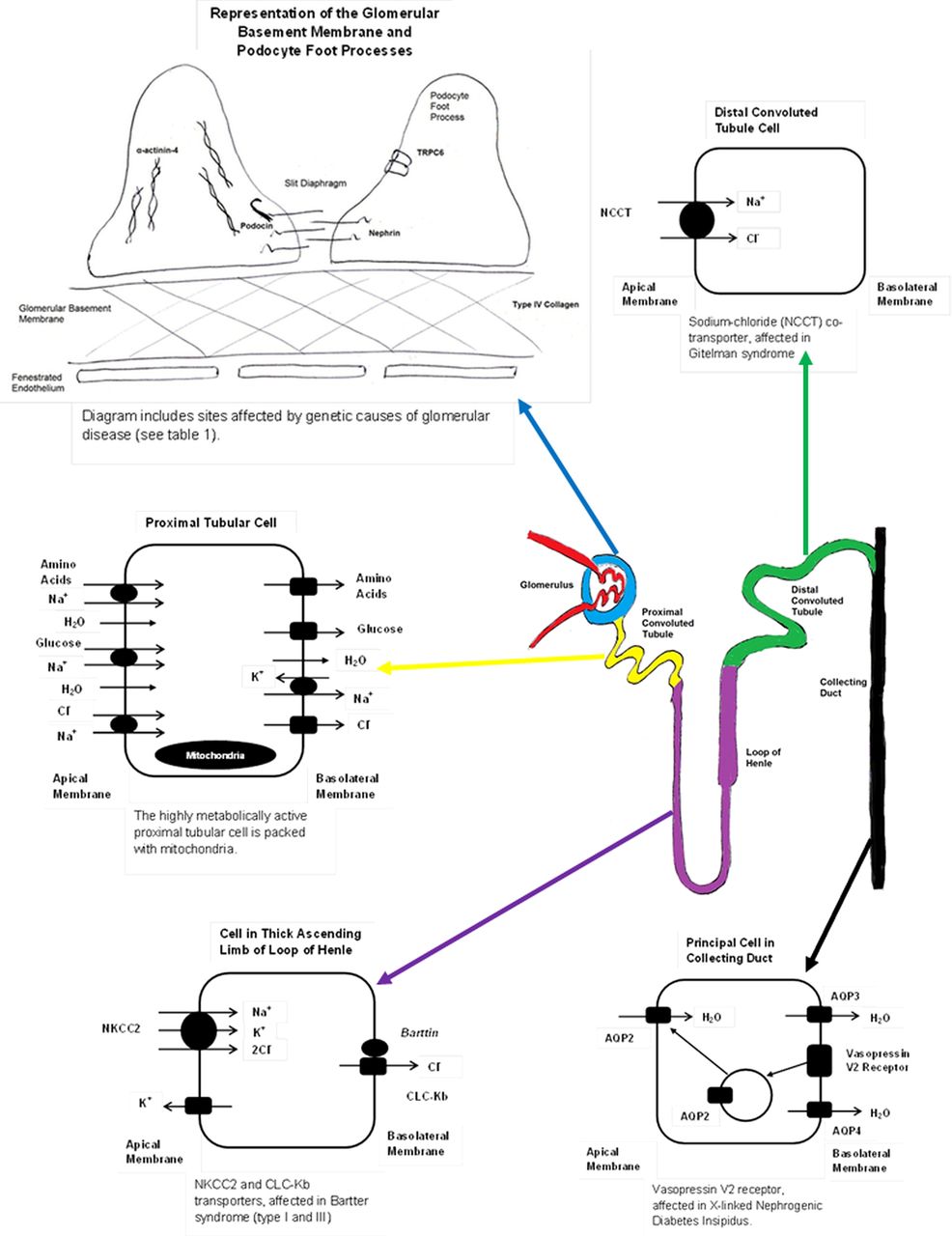
Diagram showing the nephron and the different areas affected by genetic kidney diseases mentioned in this article.
The glomerulus
A common presentation of glomerular disease in childhood is nephrotic syndrome, with the typical triad of oedema, hypoalbuminaemia and proteinuria. Proteinuria is defined as serum proteins appearing in the urine in levels above what is considered physiological. Children with nephrotic syndrome secondary to a genetic cause will present in a variety of ways, depending on the mutation and its resultant effect on glomerular function. In the most severe cases, nephrotic syndrome may be present early in the neonatal period. Congenital (Finnish) nephrotic syndrome (CNS) is the most severe type and often presents around the time of birth. Affected infants typically show the features of nephrotic syndrome within the first 3 months of life. Additionally, examination of the placenta immediately after birth will often show an abnormally large placental size. Previously, Finnish CNS was a diagnosis with a high mortality, mainly as a result of the infectious and thrombotic complications of nephrotic syndrome. We can now manage infants with Finnish CNS much more effectively, and through early nephrectomies, dialysis and renal transplantation, the mortality of this condition has greatly improved.3
Genetic studies have revealed over the past 15 years that the podocyte is a key cell in the prevention of proteinuria. There are now in excess of 15 genetic mutations that code for proteins found in the podocyte. Functionally, many of these genes affect the actin cytoskeleton of this cell showing its importance. However, there are other interesting mutations that link to cellular calcium flux4 ,5 enzyme6 and mitochondrial function.7 Also of note many of these gene products, including nephrin, are located in the ‘slit diaphragm’ between podocyte foot processes revealing this important region of the filtration barrier. Interestingly, these genetic conditions do not all present in early childhood but some can manifest in adolescence or even in adulthood.
Table 1 summarises some of the more common, well-characterised, genetic causes of albuminuric glomerular disease8 ,9 and figure 1 shows the key areas in the podocyte causing disease. The result of these mutations is disruption to the glomerular filtration barrier (GFB) and loss of albumin and other large proteins in the urine, with the subsequent complications of nephrotic syndrome. In addition many of these genetic causes of nephrotic syndrome will result in chronic kidney disease, requiring renal replacement therapy (dialysis and transplantation) in a significant proportion.
Genetic causes of glomerular disease, arranged by site affected in glomerulus (see figure 1)
The podocyte is not the only region of the glomerulus to be affected by genetic disease; in Alport syndrome, the genetic mutations affect type IV collagen, the major component of the glomerular basement membrane.10 The resultant clinical picture is one of haematuria and in many cases proteinuria, with progression to chronic kidney disease. The other important phenotypic feature of Alport syndrome is bilateral sensorineural deafness, especially in boys with the X-linked form.11 These features are due to the importance of type IV collagen fibres in the glomerular basement membrane and the cochlea of the ear. When these are mutated, there is a gradual decline in renal function and hearing, as seen in Alport syndrome. To date there has not been any convincing evidence that genetic mutations linked to the glomerular endothelium layer of the GFB causes human disease; however, there is intriguing animal data to suggest that genetic mutations in the Eps15 Homology Domain (EHD) protein family which regulate endocytic recycling in the glomerular endothelium do cause breakdown of the filtration barrier and proteinuria.12
An illustrative case of glomerular disease
A 9-year-old girl was noted to be hypertensive with a blood pressure of 149/88 at the time of an atrial septal defect repair. Clinically she was mildly oedematous on examination. Her urine dipstick showed persistent proteinuria and haematuria. Her urinary protein:creatinine ratio was 1725 mg/mmol (normal <45 mg/mmol). On blood testing, her albumin was low at 24 g/L but her renal function and all other blood tests were normal. Renal ultrasound scan was also normal. She was subsequently given 4 weeks of steroids but did not respond so she underwent a renal biopsy and this showed focal and segmental glomerulosclerosis (FSGS). Further testing found her to have a Podocin gene mutation as the cause for her steroid-resistant nephrotic syndrome and FSGS. Six years after presentation she received a deceased donor renal transplant and her nephrotic syndrome has not recurred in the transplant kidney.
The knowledge of the genetic cause for this girl's nephrotic syndrome was helpful for a number of reasons. First, it allowed her steroid therapy with all its attendant side effects to stop, as steroids will have no benefit in the presence of a Podocin mutation.13 Second, knowing that the child had a mutation is reassuring after a kidney transplant. Children with genetic mutations are much less likely to have recurrent disease than those with nephrotic syndrome who enter end stage renal failure without a genetic cause. Also it allowed clinicians to offer the patient and her family more information about the prognosis and start planning for a potential renal transplant in the future. Additionally, siblings who may be affected by the same mutation may benefit from this knowledge.
The renal tubule
The tubule is made up of three physiologically distinct functional sections: the proximal tubule, the loop of Henle and the distal tubule. A wide variety of genetic and non-genetic diseases can affect each part of the tubule and the main effect of these diseases is disruption to renal physiology.
Proximal tubular dysfunction, also known as Fanconi syndrome, has a number of genetic and non-genetic causes. A child with dysfunction of the proximal tubule will most frequently present with growth failure in the first year of life. The growth failure is a consequence of the pathophysiology of Fanconi syndrome, with glycosuria, aminoaciduria and proximal renal tubular acidosis.14 This is often accompanied by rickets as a consequence of the phosphaturia seen in this condition, and polyuria due to the sodium wasting seen in Fanconi syndrome. If proximal tubular dysfunction is clinically suspected, a simple bedside test which can be helpful is a urine dipstick; if this shows glycosuria, then proximal tubular dysfunction should be considered. One of the most important and common genetic causes of proximal tubular dysfunction in the UK is cystinosis—see the case below as an example. Cystinosis is an autosomal recessive genetic disease where there is defective excretion of cystine from lysosomes, causing accumulation. The resultant effects of this cellular defect can be seen in many different organ systems, including the eyes, nervous system and skeletal muscle, as well as the kidney.
Table 2 shows some of the common genetic causes of proximal tubular dysfunction or Fanconi syndrome. Many of these genetic diseases have extra-renal manifestations and these can be helpful when trying to decide which investigations to perform. Figure 1 shows the functions of the proximal tubule. With this in mind, it is evident that proximal tubular dysfunction will have a number of effects on renal physiology. Recent advances in molecular genetics have improved our understanding of Fanconi syndrome, and it is currently accepted that there is a unifying mechanism for all the different causes of this syndrome. This is thought to be by disrupting the supply of ATP to the highly metabolically active proximal tubular cell, reducing the activity of the sodium/potassium ATPase which maintains the sodium gradient essential for solute re-absorption in the proximal tubule.14
Genetic causes of proximal tubular defects, cystinosis, tyrosinaema type 1, galactosaemia and Lowe's syndrome cause generalised proximal tubular dysfunction (Fanconi syndrome), Dent's disease causes a degree of proximal tubular dysfunction with nephrolithiasis, while cystinuria purely causes cysteine urinary stones.11 ,14 ,20
Another presentation of proximal renal tubular dysfunction is renal stones. Children with kidney stones may present with the classic symptoms of abdominal pain and macroscopic haematuria, but these may also be absent. Other symptoms may be of urinary urgency and frequency, and rarely obstruction to urine flow.15 It is important to check a urine dipstick for microscopic haematuria and then visually inspect the urine for passed stones or grit. Metabolic abnormalities are the commonest cause of renal stones in childhood, found to be responsible for 44% of childhood renal stones in one study.16 If bilateral renal stones are seen on imaging, there is an even higher likelihood of a metabolic abnormality. All children found to have renal stones need full investigations for an underlying metabolic abnormality.16 ,17 An important stone-forming disease is cystinuria, and this is an autosomal recessive genetic disorder, caused by a defective amino acid transport protein in the proximal renal tubule. This predisposes the individual to recurrent cystine stones, see table 2 for further details. Another stone-forming genetic disease is primary hyperoxaluria; however, the reason for these children getting stones is due to a primary liver problem resulting in excess oxalate being excreted in the urine.18
Two genetic diseases which affect the loop of Henle and distal tubule are Bartter syndrome and Gitelman syndrome, respectively (see table 3). These may present in a similar way, with episodes of dehydration and intravascular volume depletion secondary to sodium loss. These children may also fail to thrive and biochemical testing may reveal hypokalaemia, hypomagnesaemia and a hypochloraemic metabolic alkalosis.
There are a number of different forms of Bartter syndrome, mainly split into the antenatal type and the classical type. The antenatal type can be evident at birth, with maternal polyhydramnios and episodes of dehydration and salt loss in the neonate. The classical type of Bartter syndrome typically presents later in childhood with recurrent dehydration and failure to thrive. The effects of the genetic mutation in Bartter syndrome can be compared to the effect of loop diuretic overdose, and the clinical features are similar. The sodium/potassium/chloride (NKCC2) co-transporter in the thick ascending limb of the loop of Henle is affected in Bartter syndrome,19 ,20 and this is also the transporter targeted by loop diuretics. Figure 1 shows the action of this transporter and the effects of dysfunction, seen in Bartter syndrome.
Gitelman syndrome is often considered a variant of Bartter syndrome and tends to present at a later age with less severe clinical features. Gitelman syndrome can be compared to overdose with thiazide diuretics. In Gitelman syndrome, there is a loss of function of the sodium chloride co-transporter (NCCT) in the distal tubule,20 ,21 the same co-transporter which is targeted by thiazide diuretics. Figure 1 shows the actions of this transporter. A useful non-invasive investigation to differentiate suspected Bartter from Gitelman syndrome is a urinary calcium:creatinine (Ca:Cr) ratio. In Bartter syndrome, there is normocalciuria or hypercalciuria with a high Ca:Cr ratio; in Gitelman syndrome, there is hypocalciuria with a low Ca:Cr ratio.
Although strictly a disorder in the collecting duct, Liddle's syndrome is another important genetic renal tubular disorder which can disrupt renal physiology. In this condition, there is a gain of function mutation in the sodium channel on the mineralocorticoid-sensitive principal cell in the distal nephron. This results in uncontrolled sodium re-absorption giving the clinical features of hypertension, metabolic alkalosis and hypokalaemia. All of these effects mimic the features of hyperaldosteronism, but in Liddle's syndrome renin and aldosterone levels are classically low.20 See table 3 for further details.
An illustrative case of proximal tubular disease
A 10-month-old boy presented with severe failure to thrive and feeding difficulties. His weight and head circumference were well below the 0.4th centile for age, and his length was just above the 0.4th centile for age. He had a fair complexion, with blonde hair and blue eyes. His urine dipstick showed glycosuria and proteinuria. On admission he had abnormal blood tests with a high creatinine of 91 μmol/L, a low sodium of 127 mmol/L, a low potassium of 1.5 mmol/L and a low bicarbonate of 10 mmol/L. Further testing included a white cell cystine level which was 1.6 (normal range 0.0–0.3). This confirmed the diagnosis as cystinosis, with resultant generalised proximal tubular dysfunction. He was commenced on the cysteamine, appropriate electrolyte, fluid and nutritional replacement with gastrostomy feeding to assist this. He is currently under regular nephrology follow-up.
Detecting this genetic defect was beneficial in treating the multiple facets of cystinosis and also the commencement of the cysteamine which has been shown to remove cysteine from cells and significantly slow disease progression.22 It also allows for screening of future pregnancies if wanted.
The collecting duct
The main role of the collecting duct is concentration of the urine; therefore, the main presenting feature of collecting duct dysfunction in childhood is a failure of urine concentration—seen classically in diabetes insipidus. Diabetes insipidus results in polyuria and polydipsia, with frequent episodes of hypernatraemic dehydration. These features can be difficult to detect clinically in childhood. Infants may present with failure to thrive and non-specific irritability. Older children may present with increased thirst, nocturia and secondary nocturnal enuresis.23 It is important to identify this condition early, as episodes of hypernatraemic dehydration can lead to adverse neurodevelopmental outcomes in affected children.24
The classical genetic cause of failure of urine concentration in the collecting duct is nephrogenic diabetes insipidus. This is a condition in which the collecting ducts are resistant to the actions of antidiuretic hormone that is released from the pituitary gland. Most frequently this is inherited in an X-linked recessive manner (see table 3). In the most common form of this condition, the affected gene is AVPR2 (the vasopressin-V2 receptor) and the result of this defect is an inability to move water channels (aquaporins) from cellular vesicles to the cell wall. This results in a lack of permeability to water in the collecting duct, so water cannot be re-absorbed from the tubule back into the circulation (figure 1). This means that the medullary concentration gradient cannot be utilised and urine cannot be concentrated, even in hyperosmolar or hypovolaemic states. This results in the clinical features of polyuria and polydipsia, with no benefit derived from treatment with exogenous anti-diuretic hormone (ADH) analogues.25
Other genetic renal conditions of note that affect the whole nephron
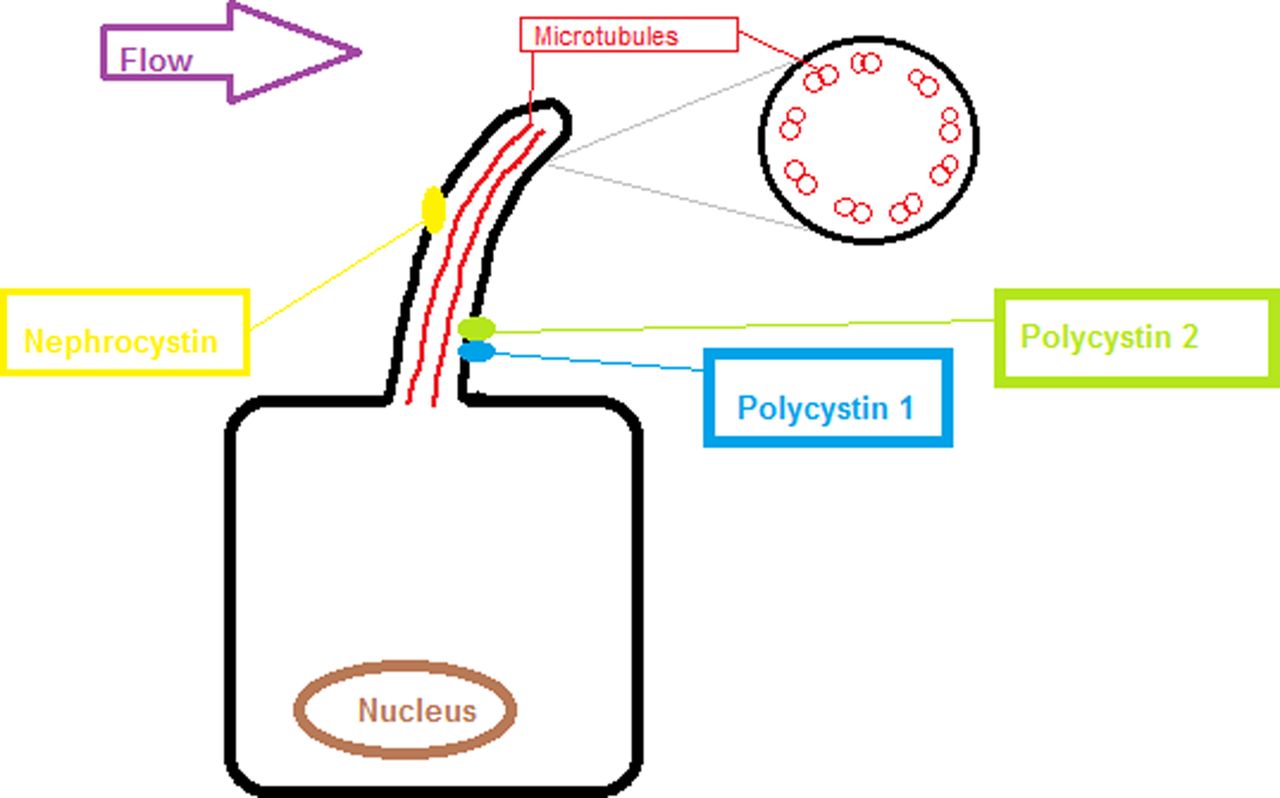
The other group of conditions that are worthy of note are the cystic renal conditions. The most common ones are nephronophthisis and polycystic kidney diseases (PKD). Genetically these are related as they are both due to genetic abnormalities affecting the primary cilia of the cell.26 They present either antenatally with large cystic kidneys, as is the case with autosomal recessive PKD. These children are often in renal failure from birth and have associated liver cystic disease. Autosomal dominant PKD classically is much slower to present and often does not become apparent until teenage years or later and commonly results in kidney failure in the 4th or 5th decade. It can also be associated with abnormalities of the circle of Willis and aneurysm formation. Nephronophthisis is interesting as it is a cystic disease associated with normal or small kidneys and multiple small cysts throughout the kidney. These children can present with renal insufficiency throughout childhood from infancy to adolescence. Some of them (approximately 10%) have extra-renal manifestation of disease including visual problems, learning difficulties and liver pathology. The genetic basis of these conditions is summarised in figure 2 and for more details of these conditions, we recommend the review by Yoder.27 The other major inherited cystic disease group worth note are those secondary to mutation of hepatocyte nuclear factor β (HNF β).28 These children present with renal cysts in childhood and there is commonly a family history of maturity onset diabetes of the young.

{kind=link}
{kind=link}
Diagram showing the primary cilium on the tubular epithelial cell in the kidney, one of the unifying features of the genetic cystic kidney diseases is that the proteins affected often subcellularly localise to the primary cilium. The proteins affected in autosomal dominant polycystic kidney diseases (polycystin 1 and 2) and nephronophthisis (nephrocystin).
Summary
Paediatricians are commonly the first port of call for patients with genetic abnormalities so it is important that we are able to recognise them and treat the patient and family appropriately. In the future, it will be possible to rapidly sequence the three billion bases in the human genome. Whether this is ethically or scientifically desirable is open to debate. However, it will lead to more understanding how the whole genome contributes to a patient's disease process.
As our understanding of genetic kidney diseases expands, the way in which we care for children with genetic kidney diseases becomes increasingly important. With knowledge of genetic mutations having an impact on prognosis and treatment, there are important decisions to be made about when and how to tell other family members, so that appropriate testing can be arranged. Joint care in consultation with clinical genetics colleagues is valuable in this setting, to provide support to the child and their family.
Within this article we have considered the various ways in which children with the commoner genetic kidney diseases may present, and the pathophysiological basis for their presentation. As more genes are discovered and the availability of genetic testing increases, clinicians will need to think carefully about which tests are selected for each individual patient.
Acknowledgments
We thank Dr Jane Tizard for her helpful review and comments on this manuscript.
References
Footnotes
-
Contributors The project was initially conceived by RJC, MM and RJC then jointly wrote and reviewed the paper.
-
Funding MM is supported by a National Institute of Health Research as an Academic Clinical Fellow. RJC is funded by the Medical Research Council as a Senior Clinical Fellow.
-
Competing interests None.
-
Patient consent None.
-
Provenance and peer review Not commissioned; externally peer reviewed.