Article Text

Statistics from Altmetric.com
Dr Tomisaku Kawasaki published a case series of 50 children in 19671 who were febrile and all had a rash, non-exudative conjunctivitis, erythema of the palms and soles of the feet, and cervical lymphadenopathy. This constellation of signs Dr Kawasaki termed “acute febrile mucocutaneous syndrome”; however the eponym Kawasaki disease has been accepted worldwide.
We consider the difficulties in diagnosis and treatment presented by this condition and examine a recently published clinical guideline of its management.
DIAGNOSIS
Kawasaki disease is a systemic vasculitis predominantly affecting children under the age of 5 years. It has a number of classic clinical features required for diagnosis.
In 1990 the American Heart Association committee on rheumatic fever, endocarditis, and Kawasaki disease2 gave the case definition that has been generally accepted—ie, a febrile illness of at least five days with at least four of the five following signs and no other reasonable cause for the findings:
-
Bilateral conjunctival injection – (there is no corneal ulceration but there may be a concomitant anterior uveitis on slit lamp examination)
-
Oral changes (erythema of lips or oropharynx, strawberry tongue due to prominent papillae, or fissuring of the lips) (fig 1)
-
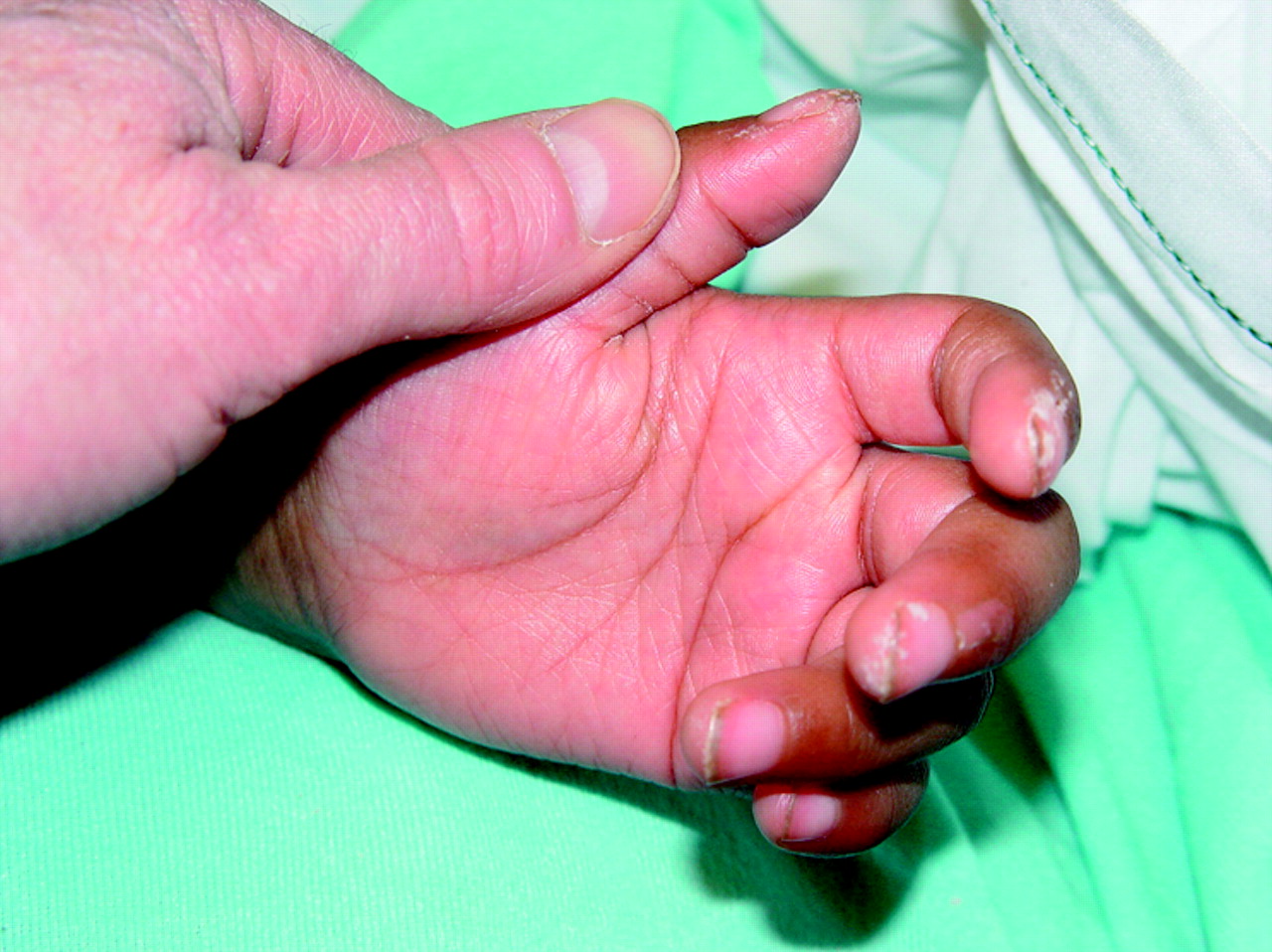
Peripheral extremity changes (oedema, erythema, or generalised or periungal desquamation); erythema is seen in the first week whereas desquamation begins about 14–21 days after the onset of the illness (fig 2)
-
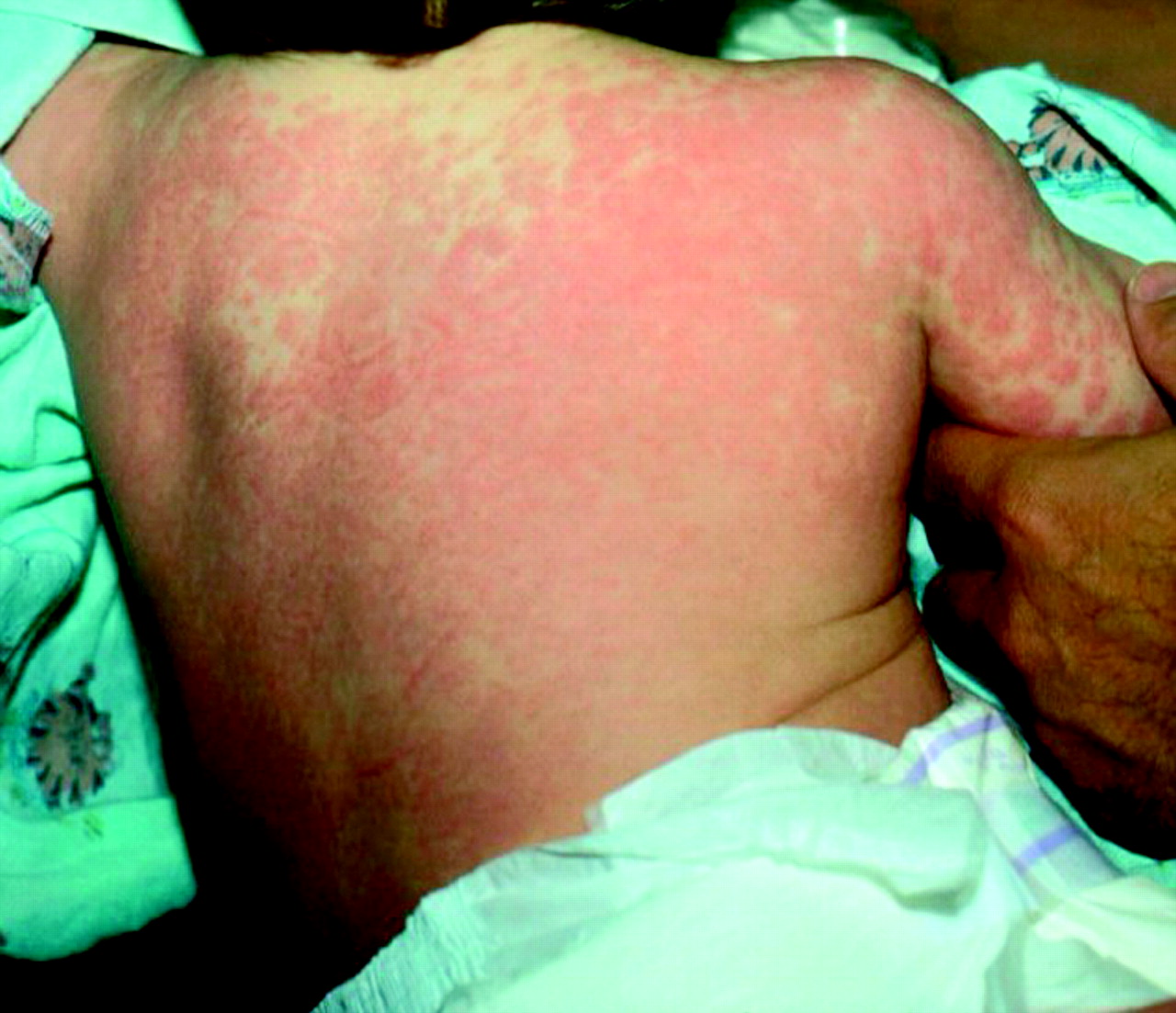
Rash – this starts in the first few days; it is often diffuse and polymorphic and lasts a week before fading. Vesicles are rarely seen; however, the rash can appear macular, maculopapular, urticarial, scarlettina or even morbilliform (fig 3)
-
Cervical lymphadenopathy is found in about 50% of cases; most often there is a painful solitary enlarged lymph gland, > 1.5 cm in diameter
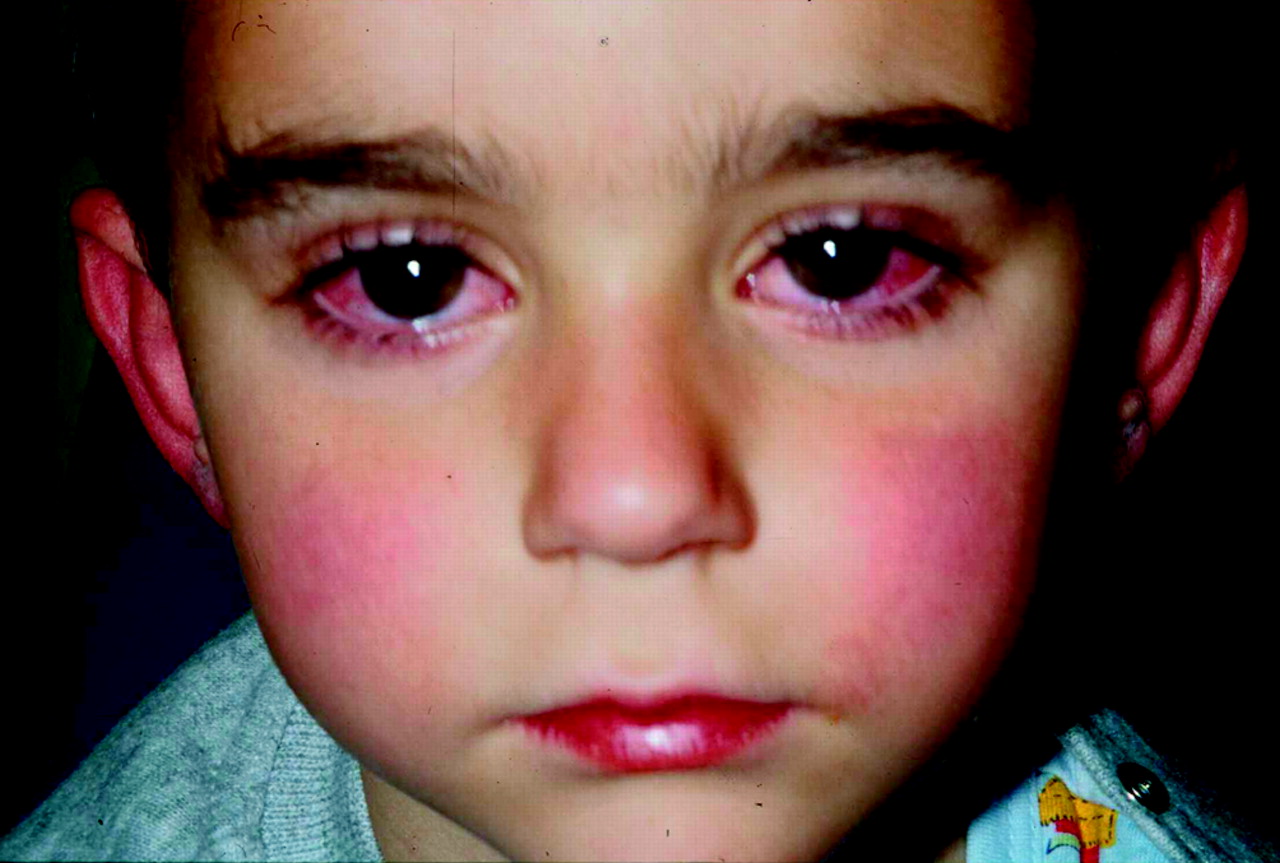
Mucous membrane changes (injected or fissured lips, redness of pharynx, strawberry-like tongue). Reproduced with permission from Falcini F, et al. Kawasaki disease in the third millineum: a syndrome still at risk to be unrecognised or underdiagnosed. Pediatric Rheumatology Online Journal 2004. (http://www.pedrheumonlinejournal.org/April/kawasaki.html)

Red swollen hand with swollen fingertip in a 4 year old girl with Kawasaki disease. © Bernard Cohen, reproduced with permission of Dermatlas (www.dermatlas.org)

{kind=link}
{kind=link}
{kind=link}
Morbilliform eruption in a 2 year old boy with Kawasaki Disease. © Peter Rowe, reproduced with permission of Dermatlas (www.dermatlas.org)
Fever is an essential feature; it is most often sudden in onset and swinging, going above 40°C. It must last at least for five days but can persist for up to a month. If coronary arterial aneurysms (CAA) are present, one of the most important complications of Kawasaki disease, then only three of the clinical features are required to clinch the diagnosis.
There are “incomplete cases” when not all of the four (three with CAA) diagnostic clinical features are present; some of these cases may evolve into complete cases.3 Some incomplete cases are diagnosed by CAA on echocardiography or at necropsy and the benefit of hindsight of the preceding clinical features.
For most children there is a subacute phase that lasts up to 30 days and a full recovery by day 50 following the onset of the illness.
EPIDEMIOLOGY AND AETIOLOGY
Kawasaki disease is the most common cause of acquired heart disease in children in the developed world. The exact cause has not yet been established but there is considerable support for it is to be due to an infectious agent causing disease among genetically vulnerable individuals.4 Kawasaki disease is most common in Japan where rates are 10 times those in the USA, and 30 times that in the UK and Australia; worldwide the annual reported incidence varies from 3.4–100/100 000.5–11 Japanese immigrants also have raised rates compared with the population of their new country, supporting the idea of a genetic influence12; siblings of index cases in Japan having an increased incidence of 8–9%.13
Children under the age of 5 years are predominantly affected,14 with a peak incidence at 9–11 months.15 There is a peak occurrence in winter and spring months.14
The true incidence is unknown as there is probably underreporting of Kawasaki disease, particularly in its mildest or incomplete form as these patients may not come into contact with medical services—for example, in 1990 five cases in the UK were recognised only post mortem.16
Elucidating the aetiology of the disease would direct treatment and provide a more rational basis for its management. Towards this aim there has been considerable focus on a bacterial superantigen toxin being the cause of Kawasaki disease over the past decade; this superantigen is believed to act in a similar fashion to the superantigen toxins of staphylococcal and streptococcal toxic shock syndromes.17–19
There are laboratory based studies that lend support to this hypothesis. One study found that the peripheral blood macrophages/monocytes (which function as antigen presenting cells (APC)) of patients with Kawasaki disease are decreased following the administration of immunoglobulin (IVIG). The APC and the T cell are implicated in superantigen disease, as the superantigen binds across the APC to the variable region of the T cell non-specifically at the V β2 region hence causing a massive upregulation of T cell activation; as IVIG has considerable benefit in treating Kawasaki disease, this would lend support to the idea of superantigen involvement in its aetiology.20
In Kawasaki disease there is a large increase in circulating B cells and fewer T cells; the effect of IVIG in vitro on peripheral lymphocytes is to decrease the percentage of B cells, increase T cells, CD4, CD8 T and CD5+ cells in acute Kawasaki disease (there is a much lesser effect with aspirin alone).21
Another in vitro study looking for the superantigen aetiology was more equivocal. In this study three colour flow cytometry was used to look at the T cell antigen receptor variable β region families from T cells of Kawasaki disease patients. These were examined pre- and post-immunoglobulin treatment and at 40 days after treatment, being compared with matched paediatric patients and one of their own healthy parents. Of all the V β families studied only VF β 2 exhibited statistical differences between pre- and post-IVIG, but the authors of this work questioned the importance of this in the underlying pathoimmunology. There was no association between V β 2 findings and T cell activation or adhesion markers, but with V β 2 abnormality both CD4+ and CD8+ abnormalities were found. The authors also suggested that patients with Kawasaki disease and cardiac involvement had a more restricted cytotoxic T cell response.22
No bacterial agent elaborating a Kawasaki superantigen toxin has been found.
DIFFERENTIAL DIAGNOSIS
The differential diagnoses of Kawasaki disease include:
-
Streptococcal infection (including scarlet fever, toxic shock-like syndrome)
-
Staphylococcal infection (such as toxic shock syndrome or scalded skin syndrome)
-
Measles, rubella, roseola infantum, Epstein Barr virus, influenza A and B, adenovirus
-
Mycoplasma pneumoniae
-
Stevens-Johnson syndrome
-
Systemic idiopathic juvenile arthritis
One of the difficulties of securing the diagnosis is that the clinical features of Kawasaki disease may appear sequentially rather than at the same time, and the feature most commonly identified is desquamation, which occurs late in the disease when cardiac complications may have occurred.
Many of the differential diagnoses can be ruled out clinically; few have a fever that persists for more than five days.23,24
The child with Kawasaki disease is very irritable and inconsolable (which may be due to an aseptic meningitis); however, this may be seen in other infections especially with measles. Another clinical sign is the presence of erythema and induration at the BCG immunisation sites as there is cross reactivity between the heat shock proteins and the T cells of patients with Kawasaki disease.25
The rash, oral and peripheral changes of scarlet fever are similar to Kawasaki disease, but the lymphadenopathy is more extensive and conjunctivitis is not seen. The rash in scarlet fever normally begins on day 2–3 of the illness, starting in the groins or axillae and rapidly spreading to the trunk, arms and legs. Seven to 10 days later desquamation occurs. The high fever associated with scarlet fever lasts 5–6 days. Scarlet fever responds readily to penicillin treatment or erythromycin for those allergic to penicillin.
Toxic shock and toxic shock-like syndromes are both associated with an ill child who may have erythema of the hands and feet, a diffuse non-specific rash over the face, trunk and limbs that desquamates, mucositis with oral involvement and non-exudative conjunctivitis. The patient needs urgent treatment with antibiotics and supportive therapy. The initial presentation of Kawasaki disease is not with shock.
Scalded skin syndrome is included as a differential diagnosis as there is a macular erythema that starts on the face and becomes more widespread; however, the epidemolytic toxin of Staphylococcus aureus (phage type II but occasionally I or III) causes bullae by separating intraepidermal layers, with the upper layers falling off. There is no mucosal involvement.
Measles mimics Kawasaki disease as there are many common features, namely the rash, non-exudative conjunctivitis, high temperature and generalised lymphadenopathy. In over half the cases of Kawasaki disease there is a solitary enlarged cervical lymph gland.
The temperature in measles may exceed 40°C but tends to fall after day 5 of the illness. Koplik spots are not seen in Kawasaki disease and the morbilliform rash of measles begins from the ears and hairline and starts to fade by day 4; after day 7 brownish staining may be seen due to capillary haemorrhage. Desquamation in severely affected cases of measles can occur but is not seen in the hands and feet.
Rubella characteristically involves the cervical, suboccipital and post-auricular glands, which may appear up to a week before the onset of the rash. The rash comprises fine pink macules that coalesce on the face and trunk, spreading to the extremities, lasting for up to five days. The temperature in children is rarely above 37.4°C.
Roseola infantum has a sudden onset of fever up to 40°C, which lasts for 3–5 days. As defervesence occurs, a generalised macular or maculopapular rash appears on the trunk and neck which lasts for 1–2 days; it may also spread to the legs and arms. Cervical lymphadenopathy is seen, the suboccipital, posterior auricular and posterior cervical nodes being enlarged. The short duration of fever and absence of mucosal involvement excludes Kawasaki disease.
Epstein Barr virus causes infectious mononucleosis that predominantly affects older children, although an anginose form affecting the tonsils is seen in preschool children, which is associated with fever and sore throat with cervical lymphadenopathy, and the clinical picture is that of acute streptococcal tonsillitis. There is not commonly a rash in this form of Epstein Barr virus infection.
Infectious mononucleosis starts with anorexia, malaise and low grade fever that lasts for 1–3 weeks. There is often notable enlargement of cervical lymph glands and splenomegaly is common. Rashes are seen in 10–15% of cases, the most common being a widespread maculopapular rash. Laboratory testing readily differentiates this condition from Kawasaki disease.
Influenza A in young children causes fever above 39°C, upper respiratory tract symptoms, and fleeting morbilliform rashes. The duration of the fever is 3–5 days at the most.
Adenovirus infection occurs mostly in children younger than 5 years and can have a number of presentations including pharyngoconjunctival fever with pharyngitis, headache, myalgia and unilateral or bilateral follilcular conjunctivitis, with exudation.
Mycoplasma pneumoniae, which may have a role in Stevens-Johnson syndrome causes upper and lower respiratory tract disease and is associated with a polymorphous rash and fever. There may also be generalised lymphadenopathy but rarely is there conjunctivitis, erythema of palms and feet or oral involvement.
Stevens-Johnson syndrome is characterised by erythema multiforme and causes erosive lesions at mucosal sites such as the conjunctivae and the oral cavity. The rash usually fades within 10 days but there is a risk of superadded infection, which may cause widespread lymphadenopathy.
Systemic juvenile idiopathic arthritis may present with swinging fevers, systemic upset, and arthritis. The arthritis may not be present at the onset of the symptoms and varies from monoarticular to more commonly polyarticular involvement. Temperatures may exceed 40°C and last for at least two weeks. There must be one or more of the following extraarticular features: generalised lymphadenopathy (painless rather than painful in Kawasaki disease), rash (classically described as a macular pink fleeting rash), hepatomegaly (not a characteristic feature of Kawasaki disease) or serositis.
Recommended investigations in combination with clinical assessment to exclude Kawasaki disease are given in table 1.26
Recommended investigations and clinical assessment to exclude Kawasaki disease
The differential diagnoses for children that do not respond to treatment with IVIG include polyarteritis nodosa, malignancy and systemic juvenile idiopathic arthritis.
Results of laboratory investigations
There may be abnormal laboratory findings such as raised white cell count with neutrophilia (> 20 × 106/ml) changing to a lymphocytosis at the end of the first week.
By day 14 there may be a hypochromic anaemia and thrombocytosis (> 1000 × 109/ml).
Erythrocyte sedimentation rate is usually raised and C reactive protein and sterile pyuria and proteinuria can be detected. None of these tests is pathognomic of Kawasaki disease.
Complications of Kawasaki disease
-
Irritability and aseptic meningitis
-
Gallbladder hydrops
-
Diarrhoea
-
Hepatitis
-
Otitis media
-
Pancreatitis
-
Myositis
-
Pericarditis and myocarditis
-
Aneurysm formation can lead to peripheral gangrene, cerebral infarction and cardiac arterial aneurysm (this may lead to thrombosis, myocardial infarction and dysrhythmia)
CARDIAC COMPLICATIONS
Cardiac involvement may be detected clinically in the second week by a pericardial rub from pericarditis, gallop rhythm, or by heart failure due to myocarditis. The most common complication of concern is CAA formation and coronary artery dilation that occurs within 6–8 weeks of disease onset. The prevalence of CAA in untreated children is 18–23%.27
CAA is detected by echocardiography; this may also reveal pericardial effusion, mitral regurgitation (myocardial ischaemia affecting papillary muscle function) or left ventricular dysfunction. Angiography is the gold standard to determine the presence of subsequent coronary artery stenosis.
Recovery from CAA is seen clinically after the first month; however, risk of death is greatest within the first 60 days from coronary thrombosis and myocardial infarction.5
Mortality rates from Kawasaki disease vary from 0.08% in Japan to 3.7% in the UK. The differences in mortality have been attributed to advances in treatments and better recognition in Japan.26
About 50% of the coronary artery abnormalities regress within five years; mild CAA (3–4 mm in size) regress within two years. CAA > 8 mm tend not to resolve.26 Patients may have long term morbidity from coronary artery scarring, developing intimal thickening and accelerated atherosclerosis.28 Evidence for this comes from a study of the response of the coronary arteries after Kawasaki disease to intracoronary injection of acetylcholine.29 This study examined the coronary luminal response to 15 μm of acetylcholine infused into either the right or left coronary arteries in 19 patients who had had Kawasaki disease. Matched control patients with congenital heart disease received the same dose. Normal vasodilation responses were seen in the controls and patients with angiographically normal vessels after Kawasaki disease; post-Kawasaki disease patients who had either persistent aneurysms or regressed aneurysms demonstrated no change or mild vasoconstriction to acetylcholine challenge. The study concluded that for patients with regressed or persistent aneurysms, the coronary arteries had residual impairment and so were predisposed to coronary atheroma development.
Early diagnosis and treatment of Kawasaki disease is important to prevent complications, reducing mortality and morbidity from this condition. The aim of treatment is to reduce the inflammatory process, so reducing the development of CAA and other vasculitic complications.
CLINICAL GUIDELINE FOR THE MANAGEMENT OF KAWASAKI DISEASE IN UK
In 2002 a clinical guideline to aid UK paediatricians provided a clear flowchart, with evidence provided to justify the recommendations, of instructions for the management of Kawasaki disease (table 2). This is an important and helpful flowchart with evidence provided. Since the publication of this guideline there has been more published evidence, relating to the use of IVIG, aspirin and corticosteroids.
Recommended guideline for the management of Kawasaki disease in the UK
Prompt recognition and treatment is needed to reduce the UK mortality figures. Highlighting that incomplete cases can be treated is important as it has been suggested that incomplete cases may be more likely to develop more coronary anomalies that patients with complete Kawasaki disease.30 The basis for selecting infants in the guideline is presumably that Kawasaki disease has a peak incidence in this age group; the authors do recommend however that all incomplete cases are treated at the discretion of the admitting clinician so that older children can be managed according to the guideline.
Aspirin
Aspirin has been considered beneficial in controlling pyrexia, conjunctivitis and mucositis.31,32 However, a Cochrane review on the use of aspirin33 in Kawasaki disease failed to find any randomised controlled trials (RCTs). One of the papers34 that did not state the method of randomisation (the author did not reply to requests for further details and so the paper was excluded) found that for patients treated with either IVIG 200 mg/kg daily for five days or IVIG 200 mg/kg/ plus aspirin 30–50 mg/kg/day for five days failed to show any difference in the incidence of CAA up to 30 days following disease onset.
The review also cites a quasi-randomised study35 that failed to show a difference between CAA in high dose (100 mg/kg) and low dose (30 mg/kg) aspirin regimens at 14 days. The higher dose was associated with a reduction in the duration of fever by 2.2 days (3.87 to 0.5 days).
Two meta-analyses were also reviewed.36,37 The Cochrane reviewers pointed out that both had methodological difficulties. In the first, none of the RCTs directly compared aspirin doses, aspirin versus placebo or IVIG, and IVIG and aspirin, and in the second retrospective and prospective RCTs comparing IVIG and aspirin had their data pooled to show that CAA was significantly lower at 30 and 60 days than with aspirin alone.
The Cochrane review concluded that until there are good quality RCTs, children with Kawasaki should continue to receive salicylate as part of their treatment regimen as there are no adverse effects reported in this group of children receiving salicylates in this dose range.
Intravenous immune gamma globulin
A Cochrane review of the effect of IVIG on the incidence of CAA was published in 2003.38 A significant decrease in new CAA in favour of treatment with IVIG compared with placebo was seen at 30 days (relative risk 0.74; 95% CI 0.61 to 0.90). If the children with CAA found at enrolment were excluded, there was an improved benefit in children receiving IVIG (RR 0.67; 95% CI 0.46 to 1.00). There was a trend towards benefit at 60 days with IVIG treatment.
There was a decrease in the number of new CAA using higher doses of IVIG; the meta-analysis of 400 mg/kg/day for five days compared with a single dose of 2 g/kg showed a significant reduction the CAA at 30 days (RR 4.47; 95% CI 1.55 to 12.86). The duration of the fever was also significant lessened with the higher dose.
There was no difference between the different types of preparation of IVIG used and no differences in the incidences of adverse effects in any group.
The conclusion of this Cochrane review was that Kawasaki disease should be treated with 2 g/kg single dose IVIG within 10 days of onset of disease, which is in accordance with the guideline. Similarly the finding that there may be benefit up to 60 days means that even children with a delayed diagnosis may benefit from treatment.
Steroids
A recent Cochrane review39 found eight RCTs that compared corticosteroids with IVIG treatment but owing to methodological differences the results were not pooled and the reviewers commented that the quality of the RCTs was poor.
Three studies showed no difference between methylprednisolone and IVIG or aspirin in the incidence of CAA (RR 0.71 to 1.33; 95% CI 0.22 to 4.97). Four studies showed an increased morbidity with prednisolone compared with IVIG or aspirin (RR 1.5 to 7.00; 95% CI 0.64 to 55.87) and one had no data.
The incidence of coronary dilation was increased in five studies in the steroid arm (RR 1.24 to 9.00; 95% CI 0.64 to 69.74), there was no effect in one and two had a slight decrease compared with placebo (RR 0.8 to 0.89; 95% CI 0.19 to 3.37).
One study showed that aspirin and methylprednisolone reduced the course of fever; two further studies showed a small effect and the remaining five studies had no data.
The reviewers found that there was a tendency to increase CAA and coronary artery dilation although steroids may have shortened the duration of fever, hence ‘we should choose steroid therapy in Kawasaki disease with more prudence’.
This indicates that better quality RCTs should be performed looking at the use of steroids in this disease and that the children who fail to respond to IVIG should be discussed with experts before starting methylprednisolone as indicated in the guideline. As seen in the differential diagnoses there are other diseases that are IVIG resistant.
Follow up
Correspondence following the publication of this guideline called into question the need for long term follow up given the limited resources in the UK National Health Service.40,41 In reply, the guideline authors42 cited evidence that long term abnormalities of endothelial function was present years after the resolution of the illness43 and called for the need to monitor these children as the long term cardiac risk is unknown.
Current evidence supports the use of the guideline in the UK, and the need for continued research into the aetiology, pathogenesis, epidemiology, and treatment of Kawasaki disease.
Acknowledgments
Thanks are due to Richmal Oates-Whitehead for providing valuable information and advice.
REFERENCES
Footnotes
-
I K Maconochie was involved as a reviewer in Oates-Whitehead R, Baumer J, Haines L et al. Intravenous immunoglobulin for the treatment of Kawasaki disease in children (Cochrane Review). In: The Cochrane Library, Issue 4, 2003. Chichester: John Wiley & Sons, Ltd; and the Cochrane review of the use of salicylate for the treatment of Kawasaki disease in children.