Article Text

Abstract
Background and aim: A role for the intestinal microbial community (microbiota) in the onset and chronicity of Crohn’s disease (CD) is strongly suspected. However, investigation of such a complex ecosystem is difficult, even with culture independent molecular approaches.
Methods: We used, for the first time, a comprehensive metagenomic approach to investigate the full range of intestinal microbial diversity. We used a fosmid vector to construct two libraries of genomic DNA isolated directly from faecal samples of six healthy donors and six patients with CD. Bacterial diversity was analysed by screening the two DNA libraries, each composed of 25 000 clones, for the 16S rRNA gene by DNA hybridisation.
Results: Among 1190 selected clones, we identified 125 non-redundant ribotypes mainly represented by the phyla Bacteroidetes and Firmicutes. Among the Firmicutes, 43 distinct ribotypes were identified in the healthy microbiota, compared with only 13 in CD (p<0.025). Fluorescent in situ hybridisation directly targeting 16S rRNA in faecal samples analysed individually (n = 12) confirmed the significant reduction in the proportion of bacteria belonging to this phylum in CD patients (p<0.02).
Conclusion: The metagenomic approach allowed us to detect a reduced complexity of the bacterial phylum Firmicutes as a signature of the faecal microbiota in patients with CD. It also indicated the presence of new bacterial species.
- CD, Crohn’s disease
- IBD, inflammatory bowel disease
- FISH, fluorescent in situ hybridisation
- OTU, operational taxonomic unit
- HSL, healthy subject library
- CPL, Crohn patient library
- PCR, polymerase chain reaction
- SSC, saline sodium citrate
- SDS, sodium dodecyl sulphate
- Crohn’s disease
- metagenome
- faecal microbiota
- phylogeny
- Firmicutes
Statistics from Altmetric.com
- CD, Crohn’s disease
- IBD, inflammatory bowel disease
- FISH, fluorescent in situ hybridisation
- OTU, operational taxonomic unit
- HSL, healthy subject library
- CPL, Crohn patient library
- PCR, polymerase chain reaction
- SSC, saline sodium citrate
- SDS, sodium dodecyl sulphate
Under certain circumstances, the indigenous gut microbes will be determinant in the onset and maintenance of inflammatory bowel disease, especially Crohn’s disease (CD), to which genetic susceptibility and disorders in mucosal immunity could also be implicated.1 Mutations in the CARD15/NOD2 gene have been associated with a higher risk of CD.2 Similarly, dysbiosis has recently been coined as a trait linked to CD.3
As most intestinal commensals cannot be cultured,4,5 genomic strategies have therefore been developed to overcome this limitation. Using classical techniques, such as temporal temperature gradient gel electrophoresis6 and single strand conformation polymorphism,7 a few studies have shown alteration in the diversity of the microbiota in CD but the precise microbial species or metabolites involved remain unknown.
The metagenomic approach, in which the microbial community is studied as a single dynamic entity, has been used to investigate complex environments such as water and soil,8,9 but has not yet been applied to the human intestinal microbiota. In metagenomics, microbial genomes are fragmented and cloned in order to obtain a single library representing the microbiome (genomes of a complex community). Whereas former researches focused on comparisons of polymerase chain reaction (PCR) derived 16S rDNA libraries, metagenomic libraries similarly permit comparison of species diversity based on a PCR independent approach but also a complete description of genes based on genomic sequence analysis and thereby a full description of functionalities of the whole ecosystem.
Here we present the first metagenomic approach to investigate the faecal bacterial composition in CD. We isolated genomic DNA directly from faecal samples of six healthy donors and six CD patients, and used the pooled DNA to construct one library for each group of subjects. The libraries were then screened for 16S rRNA genes by DNA hybridisation, and microbial diversity was determined by 16S rRNA gene sequencing and phylogenetic analysis. The metagenomic results were confirmed by using a fluorescent in situ hybridisation (FISH) method applied to individual faecal samples from the same subjects.
MATERIALS AND METHODS
Methodology
We extracted genomic DNA from faecal samples of six healthy individuals and six patients with CD. We used the pooled DNA to construct two metagenomic libraries for each group of subjects. Each library contained 25 000 clones. Two macroarrays were built after fosmid extraction and DNA spotting on nylon membranes for each library. In order to analyse microbial diversity, 16S rRNA genes were screened by DNA hybridisation and sequenced, and phylogenetic analysis was performed (fig 1).

Construction and screening of metagnomic libraries. Bacterial cells were recovered by Nycodenz density gradient centrifugation from faecal samples of six healthy subjects and six patients with Crohn’s disease in remission. High molecular weight bacterial DNA fragments (38–48 kbp) were isolated. Metagenomic DNA was then cloned into fosmids. Screening for 16S rRNA genes in the fosmid libraries was performed by DNA hybridisation on membrane nylon. All sequences were determined using four primers (ACM 008 F, ADM 330 F, SSM 1100 R, and TTM 1517 R) specific for bacteria (table 1). Assembled sequences covering approximately positions 200–1300 were selected for subsequent phylogenetic analysis. For each library, sequences were aligned using the ClustalW v1.83 program. Distance matrices were computed with DNADIST v3.6 and trees were constructed with NEIGHBOR v3.6.
Patients and samples
None of the patients or healthy subjects had received antibiotics for at least three months before sampling. The six patients with CD were five women and one man, aged 18–44 years. Patients were in remission (with a CD activity index <150). All had ileal disease; four patients had associated lesions in the duodenum, caecum, left colon, and anus (one case for each associated lesion). Two patients received no treatment at the time of sampling; two received 5-ASA (2 or 3 g/day) and two received azathioprine (150 mg/day). The six healthy volunteers were one woman and five men, aged 29–43 years.
Faecal samples were collected from the 12 subjects and were processed in less than three hours in order to preserve strictly anaerobic bacteria from oxygen. Bacterial cells were recovered by Nycodenz density gradient centrifugation10 and then washed twice with phosphate buffered saline (10 minutes at 8000 g). Cell pellets were then incorporated in agarose before gentle bacterial lysis10 and DNA digestion by the enzyme Sau 3AI.11
Library construction
High molecular weight bacterial DNA fragments (38–48 kbp) were isolated as previously described.10 Metagenomic DNA was then cloned into fosmid, a bacterially propagated phagemid vector system suitable for cloning genomic inserts approximately 40 kilobases in size, using the EpiFos library production kit (Epicentre Technologies, Madison, Wisconsin, USA), as recommended by the manufacturer (fig 1).
Macroarray preparation
Screening for 16S rRNA genes in the fosmid libraries involved extraction of the recombinant fosmids in order to remove the 16S rRNA gene of Escherichia coli used in the cloning system. Extraction was performed with a NucleoSpin 96 Flash kit, as recommended by the manufacturer (Macherey-Nagel, Hoerdt, France). Between 2 and 4 ng of each extracted fosmid was spotted on nylon membranes measuring 22×22 cm (Amersham Biosciences, Orsay, France) in a 5×5 format, using the Qbot robot (Genetix, Saint-Marcel, France); approximately 50 000 spots can be deposited on each membrane.
Macroarray probe preparation
In order to screen for 16S rRNA genes using the most universal probe, we used whole DNA directly extracted from faecal samples12 as a template to generate a mixed probe by PCR amplification. Primers used for amplification (ACM 008 F and EUB 338; table 1) generated PCR products of approximately 330 bp in length, representative of the diversity of the original ecosystem. Faecal samples were collected from the six healthy individuals used to construct the metagenomic library, and from two other healthy individuals. PCR was run with a Peltier PTC-100TM Thermal Cycler (MJ ResearchTM, Inc., Massachusetts, USA) at 94°C for 10 minutes, nine cycles: 92°C for one minute; 58°C for one minute; 72°C for one minute; and 72°C for 10 minutes. Forward and reverse primers were ACM 008 F and EUB 338, respectively (table 1). Concentrations of PCR products were evaluated on the basis of band intensities after gel electrophoresis. PCR products (30 ng) were labelled with [α-33P]dATP using the Megaprime DNA labelling System (Amersham Biosciences, Uppsala, Sweden).
Primers and probes used in this study
Macroarray hybridisation
The spotted membranes were first incubated in prehybridisation solution (6× saline sodium citrate (SSC), 1% sodium dodecyl sulphate (SDS), 10× Denhardt, 50% formamide) at 42°C for two hours. Then, a 33P-labelled probe (106 cpm/ml) and sonicated salmon sperm DNA (100 μg/ml, denatured for 10 minutes at 100°C) were added to the prehybridisation solution and incubation was continued at 42°C overnight. After hybridisation membranes were washed once in 2× SSC, 0.1% SDS at 42°C for 10 minutes, once in 2× SSC, 0.1% SDS at 60°C for 10 minutes, once in 0.2× SSC, 0.1% SDS at 60°C for 10 minutes, and once in 0.1× SSC, 0.1% SDS at 65°C for 10 minutes. Phosphor screens were exposed to the membranes at room temperature for five hours and then scanned with the Molecular Dynamics Storm system and analysed with Xdigitise software (http://www.molgen.mpg.de/~xdigitise/).
Sequences
All sequences were determined by Genoscope (Evry, France) on an ABI 3730 DNA sequencer using four primers (ACM 008 F, ADM 330 F, SSM 1100 R, and TTM 1517 R) specific for bacteria (table 1). The 16S rRNA gene was sequenced in four fragments which were then quality controlled using the PHRED v0.020425.c program and assembled with the PHRAP v0.990319 program. Assembled sequences covering approximately positions 200–1300 were selected for subsequent analysis. For each library, sequences were aligned using the ClustalW v1.83 program.13 Highly variable regions of the multiple alignment were removed, and conserved regions were selected with Gblocks,14 using parameters optimised for rRNA alignments.15 The use of this computerised method avoids the manual refinement of multiple alignments. Distance matrices were computed with DNADIST v3.616 and trees were constructed with NEIGHBOR v3.6.17 Unrooted trees were drawn with Treeview.18 We defined an operational taxonomic unit (OTU) as a cluster of 16S rDNA sequences sharing at least 98% similarity.19 A single representative faecal clone was selected for each OTU or ribotype. This clone was used as a reference sequence for calculating phylogenetic distances from other aligned sequences. Chimeric sequences were checked using the RDP CHECK_CHIMERA program (RDP-II release 8.1). The stability of branches was assessed by the bootstrap method with 1000 replicates. The term “uncultured OTUs” refers to species recovered by molecular methods only. Unidentified species (or totally novel species) correspond to microorganisms that match no available sequences in public databases (NCBI and RDP-II release 9).20 We used phylum and group level nomenclatures, as defined in Berguey’s Manual of Systematic Bacteriology (second edition) and the RDP Naive Bayesian classifier, respectively. The representative nature of our libraries was estimated using Good’s coverage formula.21
Good’s coverage was calculated as [1−(n/N)]×100, where n is the number of single clone OTU and N is the total number of sequences for the analysed sample.
Fluorescence in situ hybridisation (FISH)
FISH combined with flow cytometry was used to analyse the composition of faecal samples, targeting the 16S rRNA of three major groups of bacteria.22 The 16S rRNA specific oligonucleotide probes used in this study are listed in table 1. Probes EUB 338 and NON 338 were used as positive and negative controls, respectively. Probes Bac 303, Erec 482, and Clep 866 were used to detect the Bacteroides, Clostridium coccoides, and Clostridium leptum groups, respectively. The specificity of probe Clep 866 was optimised by using unlabelled mismatched oligonucleotides as competitors (table 1). Faecal samples stored at −70°C were fixed in paraformaldehyde and hybridised with specific probes, and positive signals were detected by flow cytometry as previously described.22
Statistical analysis
Data were analysed using the χ2 test or Student’s t test for two independent sets of samples. A p value <0.05 was considered to denote a significant difference.
RESULTS
Metagenomic libraries
Bacterial fractions from the six healthy individuals and the six CD patients were pooled to build a healthy subject DNA library (HSL) and a Crohn’s patient DNA library (CPL). Each metagenomic DNA library contained 25 000 fosmid clones. The cumulative size of the inserts spanned 2 Gbp, corresponding to approximately 500 times the size of the E coli genome (4.1×106 bp). The very low redundancy of the cloned fragments, as determined by randomly searching for cloned inserts having similar end sequences (data not shown), confirmed that major biases can be avoided by using this method. The DNA macroarrays were constructed after fosmid DNA extraction from the 50 000 recombinant clones and spotting on two nylon membranes, one for HSL and one for CPL (fig 1).
16S rRNA screening and sequencing, and bioinformatics analysis
Among the 50 000 clones, 1520 were positive for 16S rRNA sequences (650 from HSL and 870 from CPL). Each clone was sequenced with a set of four universal bacterial primers spanning the 1500 bp of the 16S rRNA gene sequence. After quality control and assembly, 1190 clones yielded sequences of more than 1000 informative base pairs suitable for phylogenetic analysis (536 for HSL and 654 for CPL).
Composition of the healthy and CD microbiota
Analysis of the 1190 clones identified 125 non-redundant OTUs (or ribotypes; GenBank accession Nos AY850400 to AY850541). According to public nucleotide databases (NCBI and RDP-II), 95 OTUs corresponded to species that are not contained in current culture collections (figs 2, 3). All of the molecular species thus recovered fell into four phylogenetic phyla: Bacteroidetes (53 OTUs; Gram negative bacteria), Firmicutes (54 OTUs; low GC Gram positive bacteria), Actinobacteria (nine OTUs; high GC Gram positive bacteria), and Proteobacteria (nine OTUs; Gram negative bacteria). Sixty six OTUs appeared completely novel, being unrelated to previously cultured microorganisms or to cloned rRNA sequences. The 89% coverage provided by the 1190 clones indicated that any new clone sequenced only had an 11% chance of corresponding to an unknown species. Hence our metagenomic libraries covered nearly 90% of the dominant species that could be cloned from the faecal ecosystem.

Phylogenetic relationships among the dominant operational taxonomic units (OTUs) identified in the faecal microbiome of healthy subjects (library based on 16S rRNA sequencing of 536 clones). The identification number (x) corresponds to the name of the metagenomic clone, which has been submitted to GenBank under the name Adhufec-x-ml for the healthy subject library (accession Nos AY850400 to AY850487). The identity of culturable bacteria and clone names are available from Genbank or RDP. When they are indicated side by side with clone a identification number (x), they correspond to the same OTU. Number of clones for each OTU is inserted in parentheses. Scale bar represents 10% sequence divergence. Bootstrap values are based on 1000 replications.
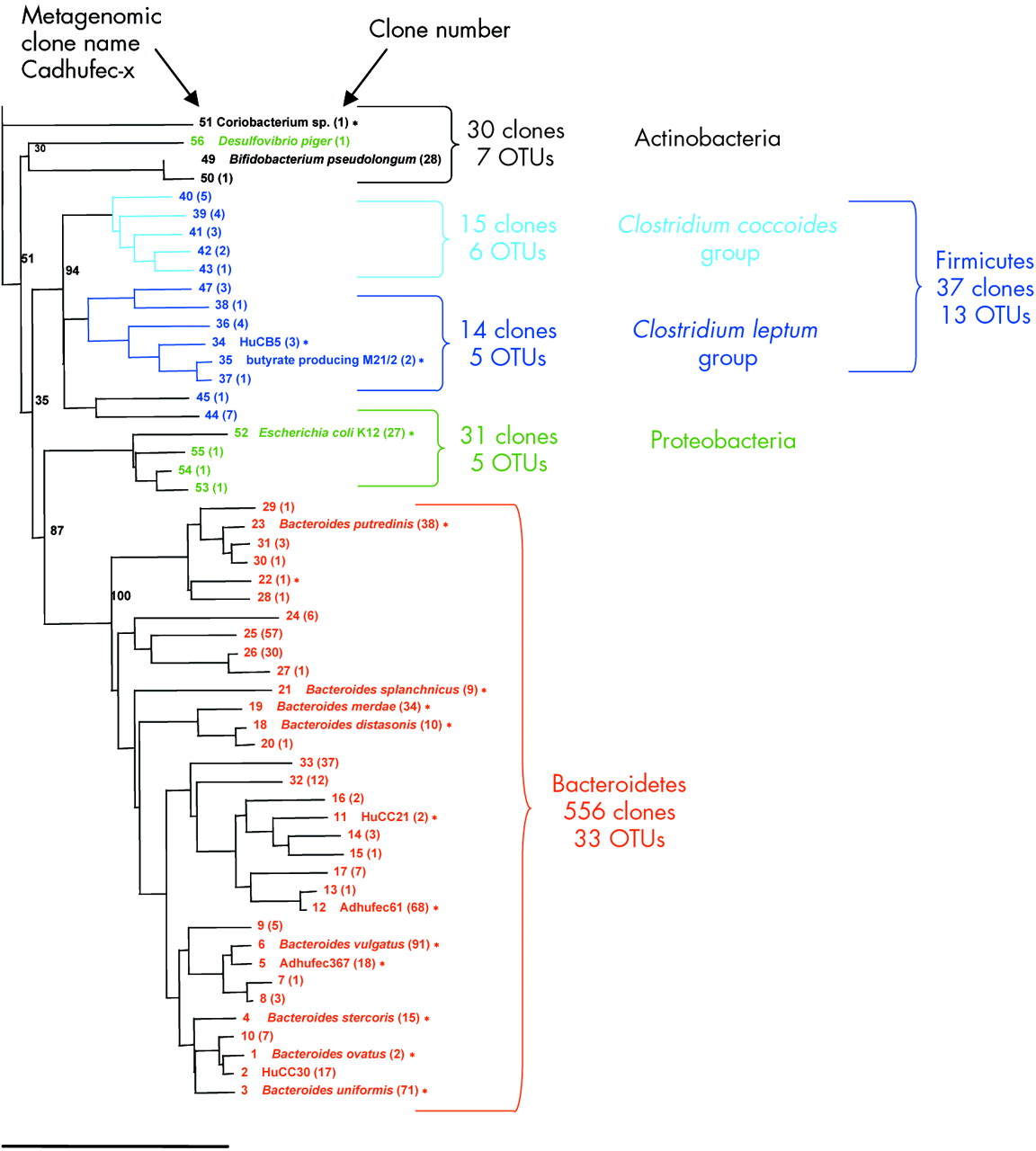
Phylogenetic relationships among the dominant operational taxonomic units (OTUs) identified in the faecal microbiome of Crohn’s disease (CD) patients (library based on 16S rRNA sequencing of 654 clones). The identification number (x) corresponds to the name of the metagenomic clone, which has been submitted to GenBank under the name Cadhufec-x-ml for the CD library (accession Nos AY850488 to AY850541). The identity of culturable bacteria and clone names are available from Genbank or RDP. When they are indicated side by side with clone numbers (x), they correspond to the same OTU. Number of clones for each OTU is inserted in parentheses. The scale bar represents 10% sequence divergence. Bootstrap values are based on 1000 replications.
Comparison of the CD and healthy libraries
Coverage values of the two libraries were not significantly different (90% and 87% for HSL and CPL, respectively). The same four dominant phyla were found in the two groups of subjects (fig 4). Bacteroidetes and Firmicutes accounted for the largest number of ribotypes, as shown by the 16S rRNA phylogenetic trees (figs 2, 3). The total number of Bacteroidetes OTUs was similar in the two groups, while differences in the Prevotella and Bacteroides fragilis subgroups were found. In addition, one “unclassified Porphyromonadaceae” species was observed only in the CD library and represented as much as 8.7% of the clones. The most striking difference was a global loss of microbial diversity in CD (88 ribotypes in healthy subjects, 54 in CD patients). This was essentially due to far fewer Firmicute ribotypes in CD patients (fig 3). Indeed, 43 Firmicute OTUs were found in healthy subjects compared with only 13 in the CD patients (p<0.005) (fig 4). The number of ribotypes shared by the two libraries was also significantly lower in this phylum (2/13, 15%) than in the Bacteroidetes group (13/33, 39%).
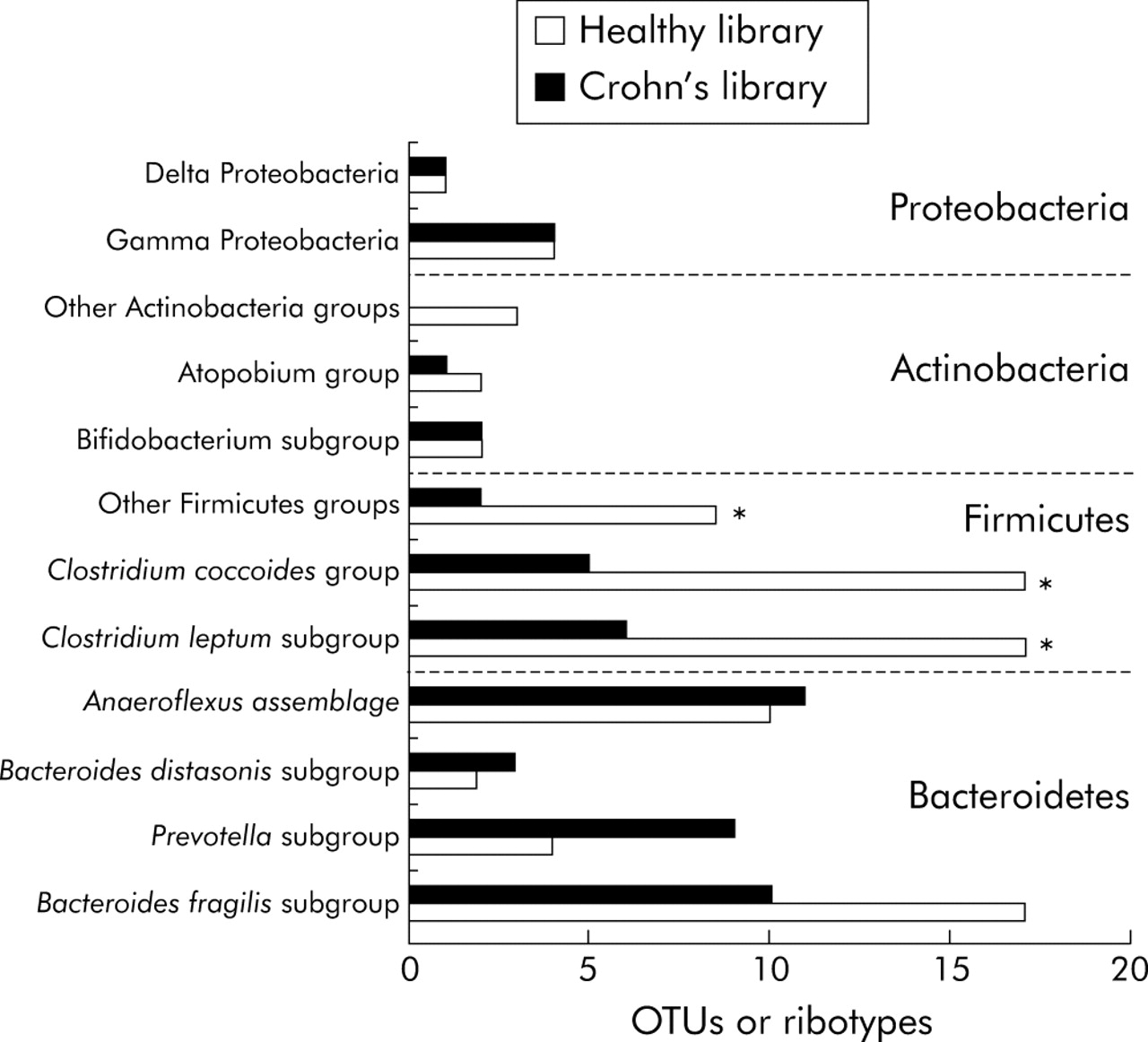
Comparison of the libraries derived from healthy subjects and patients with Crohn’s disease. The number of ribotypes belonging to each group and subgroup were compared between the two libraries. Data were analysed using the χ2 test. *p<0.025, significant difference between healthy subjects and Crohn’s disease patients.
Confirmation by FISH results
The next question to be addressed was whether this reduction in diversity in Firmicutes could be due to one patient or would it be found in every patient in the study. To address this question, we applied a FISH method to each of the 12 faecal samples individually. FISH combined with flow cytometry was used to count viable bacteria with ribosomal RNA transcription activity. Specific probes against the three major bacterial groups (table 1) revealed a significant relative reduction in the Clostridium leptum group in CD patients compared with healthy subjects (p<0.02) whereas the proportions of Clostridium coccoides and Bacteroides groups were similar in the two groups of subjects (fig 5). The results obtained by FISH and the metagenomic approach were highly consistent, showing a marked reduction in the number of active cells and in the diversity of the Clostridium leptum group in patients with CD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fluorescence in situ hybridisation (FISH) of faecal samples, coupled to flow cytometry. The 16S rRNA of the three major groups of bacteria was targeted by FISH. Faecal samples from six healthy donors and six Crohn’s disease (CD) patients were analysed independently. Mean bacterial percentages are reported for healthy donors and CD patients. Healthy subjects and CD patients were compared using the Student’s t test for two independent sets of samples. *p<0.02, significant difference between healthy subjects and CD patients for the Clostridium leptum group.
DISCUSSION
This study suggests that the faecal microbiota of patients with CD contains a markedly reduced diversity of Firmicutes. In particular, the Clostridium leptum phylogenetic group was significantly less abundant in CD patients than in healthy subjects. This observation among patients in remission suggests that it could correspond to a primary modification (that is, a modification of the faecal microbiota that may exist before the onset of the disease and/or in the absence of major perturbations caused by the inflammation process). We focused on patients in quiescence because during the active phase of the disease the microbiota is remodelled6 and alterations in its composition could be a consequence more than a cause of inflammation.
A previous analysis of amplified and cloned rRNA gene libraries from healthy volunteers (n = 3) and CD patients (n = 3)23 also indicated that the Firmicutes (Clostridium leptum and Clostridium coccoides groups) had significantly lesser complexity in CD (25 v 73 OTUs) (Irène Mangin, personal communication). These results were obtained whether the subjects were considered individually or pooled. As in the present study, the number of ribotypes in the phylum Bacteroidetes was similar in the two libraries.
Although the cause of inflammatory bowel disease remains unknown, the indigenous intestinal microbiota is considered a major if not the main trigger of inflammation, both in animal models and in humans.24 A genetically determined abnormal response to indigenous bacteria is suspected, as well as dysbiosis.3,24 However, the precise microbial species or metabolites involved remain unknown. We share the current concept that the onset of CD could be due to an altered microbiota and that a dysbiosis could enhance the risk of disease. In that respect, the reduction in proportion of one microbial group is expected to be compensated for by a greater representation of others. In the present study, the less diverse and less represented microbial species in CD patients were Gram positive anaerobic bacteria that usually account for a major fraction of the faecal microbiota of healthy subjects. These species may provide CpG DNAs with immunomodulatory activities.25 A reduced proportion of Firmicutes may also be compensated for by an increased representation of Gram negative bacteria which are known to express more proinflammatory molecules such as lipopolysaccharide. In agreement with the latter point, we observed a specific association of Gram negative species of the Porphyromonadaceae family in CD patients.
In both of our libraries, the Firmicutes comprised mainly the Clostridium leptum and Clostridium coccoides groups, along with a number of unidentified species. These two groups have been described as essential components of the human indigenous intestinal microbiota.22,26 They contain all known microorganisms, producing large amounts of butyrate, which is not only the main energy source for colonic epithelial cells27 but also inhibits proinflammatory cytokine mRNA expression in the mucosa, by nuclear factor κB activation and IκB degradation.28 Loss of butyrate producers observed here could upset the dialogue between host epithelial cells and resident microorganisms, hence contributing to the development of CD associated ulcerations.
Our results emphasise the advantages of molecular methods over culture based approaches for comprehensive description of complex microbial communities.29 Indeed, we found 69% totally novel OTUs in CD patients, compared with 34% in healthy subjects, further supporting the existence of microbial dysbiosis in CD1,23 (figs 1, 2, respectively). Known OTUs identified in this study (corresponding to previously cultured microorganisms) corresponded to species known to reside in the human29 and pig30 intestinal tract. These results are in good agreement with previous studies of human faeces using molecular approaches.23 Together, 16S rRNA sequence based studies of the human intestinal microbiota have led to the recognition of more than 50% totally novel species,31,32 suggesting that this community may be far more complex than previously thought.6
Our use of a DNA macroarray based strategy to analyse two large metagenomic libraries yielded evidence that CD may be characterised by a reduction in normal anaerobic bacterial diversity, especially among the Firmicutes, and did not support the role of a specific pathogen.
Although our study only involved a limited number of subjects (six healthy volunteers and six patients), our results were the same whether samples were pooled in the metagenomic approach or taken individually using the FISH method. Our observations point to formerly unrecognised specificities of the faecal microbiota of CD patients. Nevertheless, the number of subjects analysed was small as the metagenomic approach cannot be applied to a large set of samples. Our results should be confirmed by an epidemiological investigation focusing on the reduction in Firmicutes as well as over representation of species such as “uncultured Porphyromonadaceae”. Specific diagnostic tools could be designed to detect these. In addition, these would allow a more targeted use of antimicrobials or probiotics.
Genetic exploration of metagenomic libraries will provide access to the identification of genes and functionalities specific for CD. This is currently under investigation.
Acknowledgments
This research was supported by the French Ministry of Research. We thank C Dossat, J Castresana, P Seksik, P Robe, DM Aguilera, and the members of the CRB-GADIE INRA platform for their excellent technical assistance. We are also grateful to I Mangin, A Sghir, and D Le Paslier for fruitful scientific discussions.
REFERENCES
Footnotes
-
Published online first 27 September 2005
-
Conflict of interest: None declared.