Article Text

Abstract
This article proposes a clinical guideline for the diagnosis and treatment of Kawasaki disease in the UK based on the best available evidence to date, and highlights areas of practice where evidence is anecdotal or based on retrospective data. Future research as proposed by the London Kawasaki Disease Research Group is outlined, and clinicians are invited to prospectively enrol their suspected cases into this collaborative research project.
- Kawasaki disease
- fever
- rash
- lymphadenopathy
- Kawasaki disease
- fever
- rash
- lymphadenopathy
- CAA, coronary artery aneurysm
- IVIG, intravenous immunoglobulin
- KD, Kawasaki disease
- KDRG, Kawasaki Disease Research Group
- RCT, randomised controlled trial
Statistics from Altmetric.com
- CAA, coronary artery aneurysm
- IVIG, intravenous immunoglobulin
- KD, Kawasaki disease
- KDRG, Kawasaki Disease Research Group
- RCT, randomised controlled trial
In 1967 Tomisaku Kawasaki described 50 Japanese children with an illness characterised by fever, rash, conjunctival injection, erythema and swelling of hands and feet, and cervical lymphadenopathy.1 The mucocutaneous lymph node syndrome which he described is now recognised as Kawasaki disease (KD), and is the second commonest vasculitic illness of childhood (Henoch–Schonlein purpura being the commonest). KD is associated with the development of systemic vasculitis complicated by coronary and peripheral arterial aneurysms, and myocardial infarction in some patients.2 It is the commonest cause of acquired heart disease in children in the UK and the USA.2,3
Since the early descriptions of KD there have been 21 randomised controlled trials (RCTs), and two meta-analyses examining therapeutic interventions in KD. Despite intensive research into the illness the cause remains unknown, and although there have been significant improvements in diagnosis and treatment of children with the disease there are still a number of important unanswered questions regarding therapy. Furthermore there is still no diagnostic test available for KD.
The purpose of this article is to provide a clinical guideline for the diagnosis and treatment of KD in the UK. This guideline is based on evidence from meta-analyses and RCTs to date, and will highlight areas of practice where evidence is anecdotal or based on retrospective data. Lastly, suggestions for future research as proposed by the London Kawasaki Disease Research Group (KDRG) will be outlined.
EPIDEMIOLOGY
KD is commonest in Japan where more than 125 000 cases have been reported.3 The disease is also commoner in Japanese and other Oriental children living abroad.2 Children aged 6 months to 5 years are most susceptible, with peak incidence in children aged 9–11 months.4 Seasonal variation in the disease incidence has been reported, with peak occurrence in the winter and spring months.2 Direct person to person spread is not observed, although in Japan the disease occurs more commonly in siblings of index cases with an estimated peak incidence of 8–9% in siblings under the age of 2 years.5
Although KD is believed to be caused by an infectious agent in an immunologically susceptible individual, the causative agent remains elusive.
DIAGNOSIS OF KD
There is no diagnostic test for KD, therefore diagnosis is based on clinical criteria (table 1)2 and the exclusion of other diseases, particularly sepsis. The differential diagnosis includes toxic shock syndrome (streptococcal and staphylococcal), staphylococcal scalded skin syndrome, scarlet fever, and infection with enterovirus, adenovirus, measles, parvovirus, Epstein–Barr virus, cytomegalovirus, mycoplasma pneumoniae, rickettsiae, and leptospirosis.2,3 The differential diagnosis of IVIG (intravenous immunoglobulin) resistant KD includes polyarteritis nodosa, systemic onset juvenile idiopathic arthritis, and malignancy (particularly lymphoma).
Kawasaki disease: diagnostic criteria2
Fever of five days duration plus four of the five remaining criteria or the presence of fever and coronary artery aneurysms (CAA) with three additional criteria are required for the diagnosis of “complete” cases.3 “Incomplete” cases comprise those with fewer than the prerequisite number of criteria. Irritability is an important sign, which is virtually universally present, although not included as one of the diagnostic criteria. The exact mechanism of the irritability is unclear, but it may be related to the presence of aseptic meningitis. Another clinical sign not incorporated into the diagnostic criteria, but which is relatively specific to KD, is the development of erythema and induration at sites of BCG immunisations. The mechanism of this clinical sign is cross reactivity of T cells in KD patients between specific epitopes of mycobacterial and human heat shock proteins.6 With an increasing number of infants receiving the BCG in the UK, it is likely that this sign will become more common, and awareness of it could result in earlier diagnosis and treatment.
An important point worthy of emphasis is that the criteria may present sequentially such that a so called “incomplete” case can evolve with time into a “complete” case. Thus the diagnosis of KD must be considered in any child with a febrile exanthematous illness, particularly if it persists longer than four to five days.
Other relatively common clinical findings in KD include arthritis, aseptic meningitis, pneumonitis, uveitis, gastroenteritis, meatitis and dysuria, and otitis.7 Relatively uncommon abnormalities include hydrops of the gallbladder, gastrointestinal ischaemia, jaundice, petechial rash, febrile convulsions, and encephalopathy or ataxia.7 Cardiac complications other than coronary arterial abnormalities include cardiac tamponade, cardiac failure, myocarditis, and pericarditis.7
LABORATORY FINDINGS
KD is a multisystem vasculitic disease affecting medium sized muscular arteries, and is associated with many non-specific laboratory findings.2 Acute phase proteins, neutrophils, and erythrocyte sedimentation rate are usually increased. Thrombocytosis occurs towards the end of the second week of the illness and therefore may not be helpful diagnostically. Liver function may be deranged. Sterile pyuria is occasionally observed, and also CSF pleocytosis (predominantly lymphocytes) representing aseptic meningitis.2 Table 2 shows a list of investigations which should be performed in suspected cases of KD.
Initial investigations for suspected Kawasaki disease
TREATMENT OF KD
Treatment of KD is aimed at reducing inflammation, and preventing the occurrence of CAA and arterial thrombosis. The optimal doses of aspirin and IVIG are discussed, and the treatment of refractory KD is considered.
Aspirin
It is worthy of note that treatment of KD with aspirin alone has never been subject to a randomised controlled trial, although aspirin versus aspirin plus IVIG has been studied. Aspirin is given in the acute phase of the illness at relatively high “anti-inflammatory” doses (30–100 mg/kg/day). Following defervescence of the disease aspirin is given as an antiplatelet agent in a dose of 2–5 mg/kg once daily, the duration being dependent on findings on echocardiography.
Recent reviews have recommended the higher anti-inflammatory dose of 100 mg/kg/day in the acute phase of the illness3 on the basis of reducing duration of fever and length of hospitalisation compared with low dose aspirin (3–8 mg/kg/day).8 No such comparative data on fever and hospitalisation exists regarding high dose aspirin versus moderate doses (30–50 mg/kg/day).
Meta-analysis comparing moderate anti-inflammatory doses of aspirin (30–50 mg/kg/day) with IVIG versus high dose aspirin (80–120 mg/kg/day) with IVIG found no significant difference in the incidence of CAA between the groups.9 Currently, it is our practice to administer aspirin at a dose of 30 mg/kg/day during the acute phase of the illness, as this may be better tolerated in terms of gastrointestinal and other side effects.10 Some have advocated the use of dipyridamole in addition to moderate dose aspirin as a synergistic antiplatelet agent. The evidence for the effectiveness of this in this situation is, however, lacking.
Immunoglobulin
Early recognition and treatment of KD with aspirin and IVIG has been shown unequivocally by meta-analysis to reduce the occurrence of CAA.9,11 Moreover, the prevalence of coronary artery abnormalities in KD is highly dependent on total IVIG dose, but independent of aspirin dose.9 Treated with aspirin alone, 20–40% of children develop CAA.2,9,11 Combined therapy with aspirin and high dose IVIG given as a single infusion reduces the occurrence of CAA to 9% at 30 days, and 4% at 60 days after the onset of the illness.11 The prevalence of CAA is inversely related to the total dose of IVIG,9 2 g/kg of IVIG being the optimal dose, usually given as a single infusion.9,11 Meta-analysis of randomised controlled trials comparing divided lower doses of IVIG (400 mg/kg/day for four consecutive days) versus a single infusion of high dose IVIG (2 g/kg over 10 hours) has clearly shown the therapeutic benefits in the prevention of CAA with the latter regimen.11 One important practical point, however, is that infants who have cardiac compromise may not be able to tolerate the fluid challenge associated with the high dose single infusion, and consideration of divided doses given over several days may be appropriate for this patient group.
IVIG treatment should be started early in the disease, preferably within the first 10 days of the illness.9,11 Importantly, however, clinicians should not hesitate to give IVIG to patients who present after 10 days if there are signs of persisting inflammation.
Not all patients respond to a single dose of IVIG, and some require a second dose. It has been observed that those children who received IVIG very early in the illness may require a second infusion of IVIG for primary treatment failure or disease recrudescence.12 Thus, the timing of IVIG administration appears to be important, although this latter point should not dissuade clinicians from giving IVIG before day 5 of fever if the diagnosis of KD is suspected.
Lastly, one question that remains unanswered is whether the type of IVIG administered is important, perhaps as a result of the presence of antibodies to epitopes derived from different donor pools. There are currently no data to suggest that any particular preparation is superior.
The role of corticosteroids in KD
The use of corticosteroids in KD remains controversial. Unlike other vasculitic conditions, where corticosteroid remains the mainstay of treatment, an early study suggested that the incidence of CAA was greater in those treated with prednisolone compared with those treated with aspirin alone.13 Other workers have suggested that the use of pulsed methylprednisolone is effective in treating patients with KD resistant to IVIG.14 We believe that there is a role for corticosteroids in patients with KD refractory to treatment with IVIG,15 and also for those patients who may refuse IVIG because of religious beliefs, such as Jehovah's Witnesses. We suggest, however, that treatment of refractory KD with corticosteroids should be undertaken in specialised centres.
VACCINATION POST KAWASAKI DISEASE
There are two important issues relating to vaccination following an episode of KD. Firstly, IVIG can block replication of live viral vaccines and subsequent actively acquired immunity.16,17 Consequently it is currently recommended that live vaccines be deferred for at least three months following treatment with IVIG.18 Secondly, an important question remaining unanswered is the safety of live or other vaccines in children recovering from KD. It has been our observation that some autoimmune disease states, including the systemic vasculitides, flare in response to live and non-live vaccine preparations. It is therefore our policy to defer immunisation with all vaccines for at least three months following an episode of KD.
CARDIAC COMPLICATIONS OF KD
Echocardiographic and cardiac angiographic data indicate that 20–40% of untreated KD patients develop coronary artery abnormalities. Approximately 50% of these lesions regress within five years, and in most with mild CAA (3–4 mm) regression occurs within two years.2 Giant aneurysms (>8 mm) are unlikely to resolve, and some may develop stenosis with risk of coronary thrombosis, myocardial infarction, and death. In 1993, a report from the British Paediatric Surveillance Unit indicated a mortality rate of 3.7% in the UK for KD.19 Current mortality rates reported from Japan are much lower at 0.14%.3 The reasons for this difference include improved therapy, and better case recognition. Long term sequelae may include the early development of coronary atherosclerosis.20
All patients with KD should undergo echocardiography on diagnosis and six to eight weeks after the onset of the disease.2 We also advocate an intermediate echocardiograph at 10–14 days of disease onset to pick up any missed pathology. Additionally, it is our practice to perform echocardiography at least weekly (and occasionally 48 hourly in the acute stages with ongoing active inflammation) for those who develop CAA to monitor aneurysm size progression, or the development of thrombus formation. Long term aspirin at 2–5 mg/kg/day is recommended for those with persisting aneurysms on echocardiography.3 This can be discontinued if the aneurysms resolve. Depending on the size of the aneurysms, electrocardiography, and echocardiography performed 6–12 monthly is recommended.
Patients with aneurysms greater than 8 mm may require stress testing and possibly coronary angiography to identify stenotic lesions. Most experts recommend the addition of warfarin to aspirin therapy for those with giant (greater than 8 mm) coronary aneurysms,21 although randomised controlled trials supporting this practice are lacking. If warfarin is to be commenced, it is imperative to cover the initial warfarinisation period with intravenous heparin to counteract the paradoxical prothrombotic state which can occur soon after starting warfarin in some patients.22 More recently, it has been suggested that platelet glycoprotein IIb/IIIa receptor blockade therapy may be a useful addition to the therapeutic armamentarium for those with giant aneurysms.23
Limitation of strenuous activity is recommended in all patients with giant coronary aneurysms and/or stenoses. Some patients with coronary artery stenoses may require surgical revascularisation. In those patients who develop myocardial infarction, treatment with streptokinase or tissue plasminogen activator is indicated.2
A rare but serious complication of KD is the development of peripheral ischaemia and gangrene.2 This particular complication is associated with peripheral arterial aneurysm formation, particularly axillary aneurysms. Treatment with thrombolytic agents, anticoagulants, and intravenous prostacyclin may be indicated in such patients.2
CLINICAL GUIDELINE FOR THE MANAGEMENT OF KD
Based on the evidence available to date, we recommend the clinical guideline shown in fig 1 for the management of KD in the UK. We propose that complete cases are treated, and additionally incomplete cases are treated at the discretion of the admitting clinician. We suggest, however, that clinicians should have a lower threshold for treatment of incomplete cases under the age of 1 year. For older children with incomplete KD, the decision to treat will be influenced by factors such as the small but theoretical risk of blood borne virus infection from IVIG. Lastly, we recommend yearly lifelong follow up of all children following an episode of KD. The purpose of this is to document the blood pressure, and provide general advice regarding other cardiac risk factors.
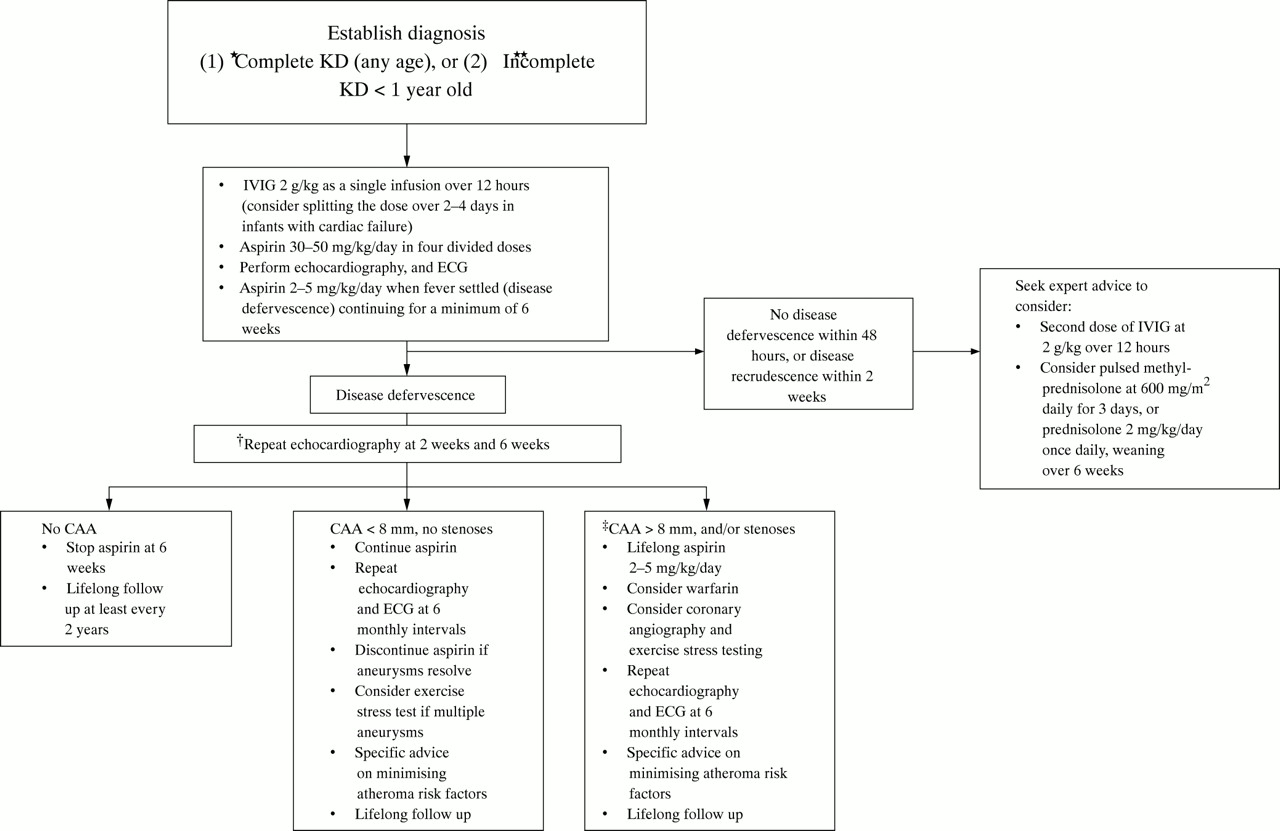
{kind=link}
Recommended clinical guideline for the management of KD in the UK. *Treatment can be commenced before full 5 days of fever if sepsis excluded; treatment should also be given if the presentation is >10 days from fever onset. **Incomplete cases >1 year old treated at discretion of clinician—seek expert advice. †Refer to paediatric cardiologist. ‡Other specific interventions such as positron emission tomography scanning, addition of calcium channel blocker therapy, and coronary angioplasty at discretion of paediatric cardiologist.
AREAS FOR FUTURE RESEARCH AND THE KAWASAKI DISEASE RESEARCH GROUP
The KDRG is made up of the principle research groups with an interest in KD in the London area. The main objectives of the group are to examine the aetiopathogenesis and changing epidemiology of KD in the UK. Specific areas of research will include: (1) the epidemiology including incidence, management, complication rate, and case fatality rate; (2) investigation of superantigen dependent or independent pathways of T cell activation in the acute and convalescent phases of the illness24–26; (3) the investigation of a viral aetiopathogenesis, in particular herpes viruses, using degenerate polymerase chain reaction methodology27; (4) investigation of host genetic determinants of susceptibility and outcome in KD5; (5) correlation of coronary and/or peripheral arterial involvement with clinical presentation; (6) evaluation of the potential of circulating endothelial microparticles as a laboratory diagnostic test and predictor of those at risk of coronary arterial aneurysm formation28,29; (7) the examination of B lymphocyte homing receptor expression of circulating antibody secreting B lymphocytes in acute and convalescent KD.
Multiregional and local research ethical committee approval has been granted in the London area. If clinicians wish to enrol a patient for research, the first step is to contact a member of the KDRG (see below). If not previously received, the appropriate information and consent forms will be faxed to the referring clinician, and advice given regarding the collection and handling of blood specimens and swabs. Samples must be collected prior to administration of IVIG, hence the KDRG bleep holder is available 24 hours/day, including weekends.
A secondary aim of the group is to provide a centralised advisory service for general paediatricians, initially within the London area, but ultimately nationwide. Clinicians are therefore invited to contact key nominated clinical representatives of the KDRG with suspected complete or incomplete cases of KD, prior to commencement of therapy with IVIG. Permission to enrol the patient into the KDRG study will be sought, and telephonic advice given if requested. The aircall number to contact (24 hours) is: 08700 555500; ask for pager number 845031 and leave a message.
REFERENCES
Linked Articles
- Miscellaneous