Article Text

Statistics from Altmetric.com
Early phase trials provide crucial information about new medicines that allow them to be taken forward into larger confirmatory studies. Paediatric early phase studies are becoming more common, particularly in the era of precision therapy. There are almost 600 active paediatric phase I/II trials listed on clinicaltrials.gov. Conventionally, early phase dose-escalation trials use rule-based designs such as the 3+3 to guide dose decisions. A trial is considered to have a rule-based design if predefined rules are used to guide decisions to escalate, continue or de-escalate based on the observed toxicities at the current dose.
Though it is well established that model-based design is generally superior to rule-based design, its uptake remains low. Trials with a model-based design use statistical models to guide decisions on which dose levels to give the next patient(s), based on the targeted toxicity level and previous information. In this article, we review one of the most commonly used model-based designs, the continual reassessment method.
The goal of any phase I study is to find a recommended dose of a new therapy to advance in subsequent studies. This can be defined in different ways; one of the most common being the maximum tolerated dose (MTD). One key question is: ‘What level of toxicity is acceptable based on the expected benefits of treatment?’ The MTD is defined as the dose expected to cause a degree of medically undesirable dose-limiting toxicity (DLT) in an acceptable specified proportion of participants. The latter is referred to as the target toxicity level (TTL). This level will differ depending on the expected benefits of the treatment as well as the severity of the expected toxicity for the specific intervention. This relationship is illustrated in figure 1.
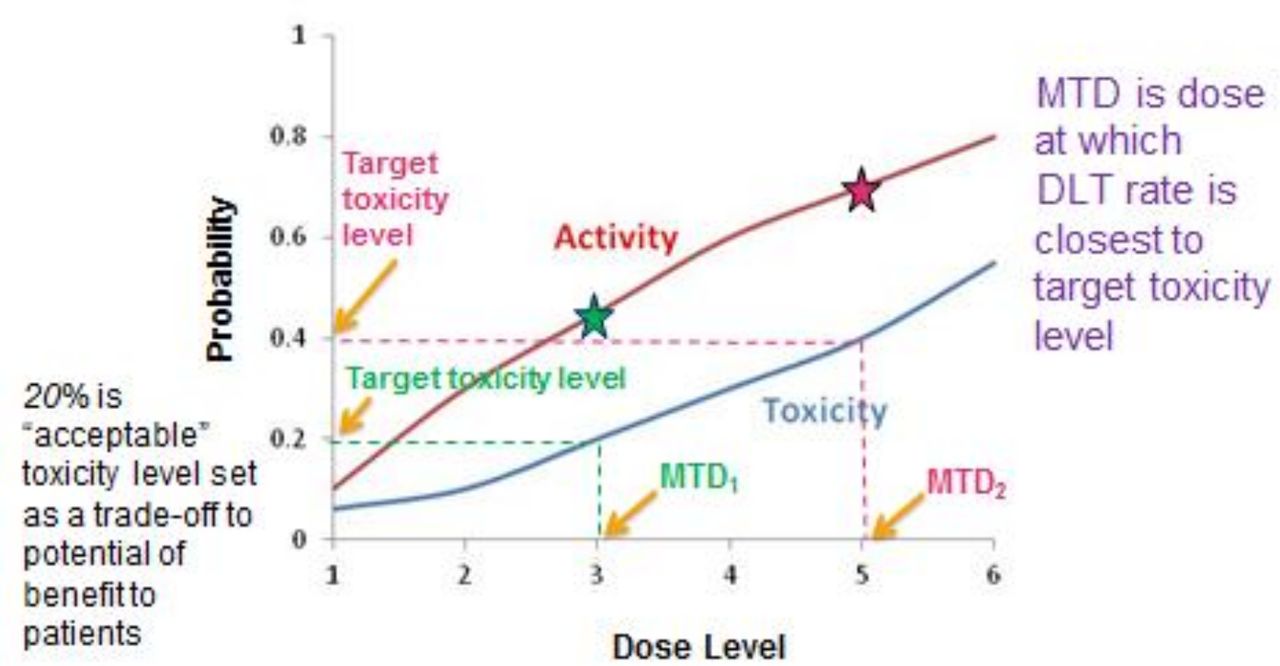
An illustration of the relationship between dose level and the probability of toxicity and activity. A target toxicity level (the proportion of patients in which it is considered acceptable to have medically undesirable side effects) of 20% will result in a MTD at dose level 3 (MTD1), which gives an activity rate (the proportion of patients who demonstrate efficacy) of 45%. A target toxicity level of 40% will result in a MTD at dose level 5 (MTD2), which gives an activity rate of 70%. The stars indicate the activity at dose levels 3 and 5. In this example, as the dose increases, the activity increases but so does the toxicity. DLT, dose-limiting toxicity; MTD, maximum tolerated dose.
With rule-based 3+3 design, cohorts of three patients are treated at each dose level and decisions on escalation or de-escalation are based on the number of patients with DLTs at the current dose. If none out of three patients experience DLTs, the dose is escalated to the next level. If one out of three DLT is observed, three further patients will be recruited and escalation happens if no further DLT is observed. If at least two out of three DLTs are observed, the dose is de-escalated to the next level. Depending on factors such as the number of dose levels and the starting dose, many patients may receive subtherapeutic doses without any toxicities. Other drawbacks are highlighted in table 1.1
Summary of some key features of rule-based and model-based designs1
What is the continual reassessment method?
The continual reassessment method is a form of model-based trial design that was first proposed in 1990 to obtain the MTD.2 Unlike rule-based approaches, a continual reassessment method uses a statistical model to estimate the relationship between dose and DLT risk. With a Bayesian approach, it integrates accumulated observed data in the trial as well as prior information from clinicians and past studies, to recommend a dose with estimated DLT risk closest to the TTL to the next cohort/patient. The model learns as the trial progresses as the data from every patient already enrolled is included to recommend the best MTD estimate for the next patient. Since its introduction, numerous modifications have been proposed to improve safety such as increasing the size of dosing cohorts and reducing the chance of large increases in dose in the early stages of a study.3 Continual reassessment method designs are particularly attractive in paediatric trials as it is exceptionally rare for a drug to be trialled in children prior to similar studies in adults. The toxicity seen in adults can be used to estimate the likely toxicity across the tested doses in children to inform an appropriate starting dose and the statistical model. In practice, if we observe patients experience DLT at the current dose, the model will assess if it has underestimated the risk of DLTs and may then reduce the dose for the next cohort if so. If patients do not experience DLT, the opposite may occur. The trial continues until predefined stopping criteria are met such as a prespecified maximum number of enrolled patients or sufficient patients being dosed at the proposed MTD.4 5
Consider figure 2 from the VIOLA Trial (A trial of combined azacitidine and lenalidomide salvage therapy in patients with acute myeloid leukaemia and myelodysplasia who relapse after allogeneic stem cell transplantation). The VIOLA Trial was a phase I study looking at the treatment of adult patients with relapsed acute myeloid leukaemia postallogeneic stem cell transplant. The prior curve shows that the study team initially overestimated the risk of DLTs. As the trial progressed and more patients were treated without DLTs, the model reduced its DLT risk estimates, resulting in the updated curves shown.

{kind=link}
{kind=link}
This figure shows the evolution in the estimates of the probabilities of DLTs through successive patient cohorts in the VIOLA Trial which used a continual reassessment method design. The dose-toxicity curves reduced through the first four cohorts as no DLTs were observed before increasing again in cohorts 5 and 6 when DLTs occurred. The MTD was declared at dose level 3 as it had an estimated DLT risk closest to the target toxicity level of 20% (adapted from Craddock et al. 7). DLT, dose-limiting toxicity; MTD, maximum tolerated dose.
A good example of a paediatric trial which used the continual reassessment method is a trial of selumetinib in patients with recurrent or refractory low grade glioma.6
Benefits of continual reassessment method over rule-based designs
A large number of studies have compared continual reassessment method design to rule-based designs and a number of benefits have been identified, including the ability to identify the recommended phase II dose more accurately. One key advantage is the flexibility to accommodate potential deviations from the plan as seen in the VIOLA Trial.7 Key features are summarised in table 1.1 8 9
Concerns about continual reassessment method trials
There are some concerns about continual reassessment method studies that currently limit their use. All model-based designs account for less than 10% of current phase I studies. They are undoubtedly more complex to design/run, requiring active input from a trial statistician. This can increase the upfront costs prior to securing funding which can deter some clinicians.10
While most early phase researchers are readily familiar with the 3+3 design which is simple and easy to implement, there can be perceived fears around the ‘black-box’ of model-based designs. However, it is straightforward to make the ‘black-box’ transparent using the concept of ‘Dose Transition Pathways’ to understand possible courses of action in a trial that implements a model-based design—both at the design stage and the conduct stage.11
Some researchers have raised concerns about the safety of continual reassessment method trials. As highlighted in table 1, continual reassessment method studies are safer and more ethical than rule-based designs by treating more patients at a dose more likely to be effective while protecting them against excessive doses. Safety mechanisms can be built-in to limit risks such as increased dose cohorts and to avoid skipping of untried doses in escalation. Crucially, these safety mechanisms do not reduce the flexibility or efficiency of the studies. Furthermore, the observation of safety data from the first patient in a study may be required before any other patients can be dosed in that cohort due to several past phase I trial disasters.5 10 11
Conclusion
Early phase clinical trials are essential components of evidence-based medicine. Due to relatively small patient numbers, it is vital that trials are designed in the most efficient way possible. Continual reassessment methods have been shown to have many advantages over conventional trial designs including efficiency and efficacy for patients. Despite this, uptake remains low with relatively unfounded fears about the complexity of designing and running them.
Footnotes
Twitter @ChristinaBYap
Contributors GCM wrote and revised the initial manuscript. CY wrote, reviewed and revised the manuscript prior to submission. Both authors approved the final manuscript.
Funding CY is funded by Cancer Research UK Grant No. C22436/A15958.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.