Article Text

Abstract
The descriptive term of cerebral palsy encompasses the largest group of childhood movement disorders. Severity and pattern of clinical involvement varies widely dependent on the area of the central nervous system compromised. A multidisciplinary team approach is vital for all the aspects of management to improve function and minimise disability. From a medical viewpoint, there are two pronged approaches. First a focus on developmental and clinical comorbidities such as communication, behaviour, epilepsy, feeding problems, gastro-oesophageal reflux and infections; and second on specifics of muscle tone, motor control and posture. With regards to the latter, there is an increasing number of available treatments including oral antispasticity and antidystonic medications, injectable botulinum toxin, multilevel orthopaedic and neurosurgical options and a variety of complementary and alternative therapies.
Statistics from Altmetric.com
Introduction – the context
In 1843, William Little, an Orthopaedic Surgeon, first linked the long-term stiffness and contractures he saw in some children to the damage inflicted on their developing infant brains – ‘cerebral deformities’. Then in 1889, William Osler published on ‘The Cerebral Palsies of Children,’ delineating specific patterns of childhood movement disorders. ‘Cerebral’ – of the brain, ‘palsy’ – a disorder of movement and posture – the descriptive term stuck.
Over the years, there have been attempts to reorganise the descriptive term, without gaining an overriding consensus on exactly what it means. To attempt a full stop (or at least a colon), following recent work from the Study of Cerebral Palsy in Europe (SCPE)1 and the American Academy of Cerebral Palsy (AACPDM), an international group led by Rosenbaum and Bax, gave us the most current unifying definition:
Cerebral palsy (CP) describes a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing foetal or infant brain. The motor disorders of CP are often accompanied by disturbances of sensation, perception, cognition, communication and behaviour; by epilepsy and by secondary musculoskeletal problems.2
This manages to encapsulate the functional problems encountered by the child, family and clinician. We can do little about the initial ‘disturbance’ so our multidisciplinary management focuses on these comorbidities and on minimising the impact of secondary deformities.
Basic science, pathophysiology and epidemiology
The site of any central nervous system impairment leads to the specific clinical signs and functional symptoms of the individual's disability. These in turn compound problems with integration into society – handicap. That's the sum of physical neurodisability and the basis of our management; try to minimise the disability and handicap. To understand impairment and how to manage sequelae, we have to start with a review of brain development, keeping it brief and simple.
Sperm plus egg = us. Cellular replication forms the blastocyst, then the morula, the ball of cells which differentiates into the three basic embryological subtypes, endoderm (centre=organs), mesoderm (middle=musculoskeletal system) and ectoderm (surface=skin and nerve). This doughnut then squashes down to form a disc.
By day 16, one side of this becomes the neural plate, down the centre of which the neural groove forms. This then rolls up and closes to form the rostral and caudal neuropores. Up the top end, massive differentiation and convolution occurs to form the primitive fore, mid and hind brain. By the time we are born the brain is over 10% of our total body weight (~400 gm), further synaptic connection, supportive glial cell formation and myelination lead to increased rapid growth and development. By age 3, the average brain weighs 1.2 kg, close on adult size.
The most rapid phase of central nervous system growth occurs from inutero, until about 5 months after we are born. During this stage, the brain is susceptible to malformation, infection, damage and disruption of its emerging blood supply, which, in turn, starts developing in the first trimester of pregnancy on the front and back surface of the brain and migrates centrally and downwards to the deeper structures.
Therefore, in the immature brain, the area most susceptible to damage from disruption to the blood flow is the central ‘watershed’, ‘periventricular zone’ of the motor cortex – the area furthest from the large blood vessels. However, by full term, the blood supply to the brain is much more robust and if hypoxic damage occurs at this stage it is initially in the cells that have the highest metabolic need – the basal ganglia.
Beyond infancy, these basal ganglia are the ‘locomotor driving system’ of our motor control, where reflex patterns of movement are unconsciously co-ordinated. Fluid motor patterns are initiated by desire and anticipation of function and are modified by the inputs such as the peripheral sensory interaction, and vision and balance – the equilibrium and adaptive systems. At this point, intracerebral reflex loops organise the descending upper motor control of the peripheral lower motor neurons via, among others, the corticospinal tracts.
It is obviously a lot more complicated, not least because of the tremendous amount of potential tract reorganisation that can occur with damage – ‘neuroplasticity’. There has been considerable research around the basic science of the projection and organisation of motor tracts during early neurological development; outlining the process of functional cortico-spinal innervation from all ipsi and contralateral areas of the brain and the subsequent pathway regression.3
Yet, to put it simply, when this complex motor system is impaired, the fluidity of control breaks down, leading to the ‘non-progressive movement disorder’.
In summary, anything that can cause non-progressive lesions of the central nervous system can lead to a movement disorder. Developmental structural abnormalities and in particular disruption in blood flow, pre, peri and postterm lead to different physical impairments and thus different clinical patterns.
With such a myriad of potential causes, ‘CP’, as a descriptive umbrella, is by far the commonest physical neurodisability during childhood and as such has a massive socio-economic impact.4 Overall prevalence in Europe, America and Australasia is fairly stable at 2–3 per 1000 live births, though in susceptible premature infants it rises to up to 100 per 1000 in those born before 28 weeks gestational age.1
Severity, motor pattern and distribution of CP
There is a massive variance in clinical presentation and range of disability observed in individuals with ‘CP’. The impact of the lesion depends on its site and severity and our management differs with the level of background functional ability.
Subdivision of the movement disorder is dependent on the distribution, severity and motor pattern observed; as well as by the accompanying comorbidity.
Distribution
Classification systems focus on the pattern of trunk, limb and bulbar involvement. There seems to be increasing agreement on using the terms unilateral or bilateral, as outlined in the SCPE; with less emphasis on hemiplegia (unilateral), diplegia (leg involvement>arms) and quadriplegia (all four limbs), though these terms are still frequently used.
Severity of the motor difficulty is assessed as follows:-
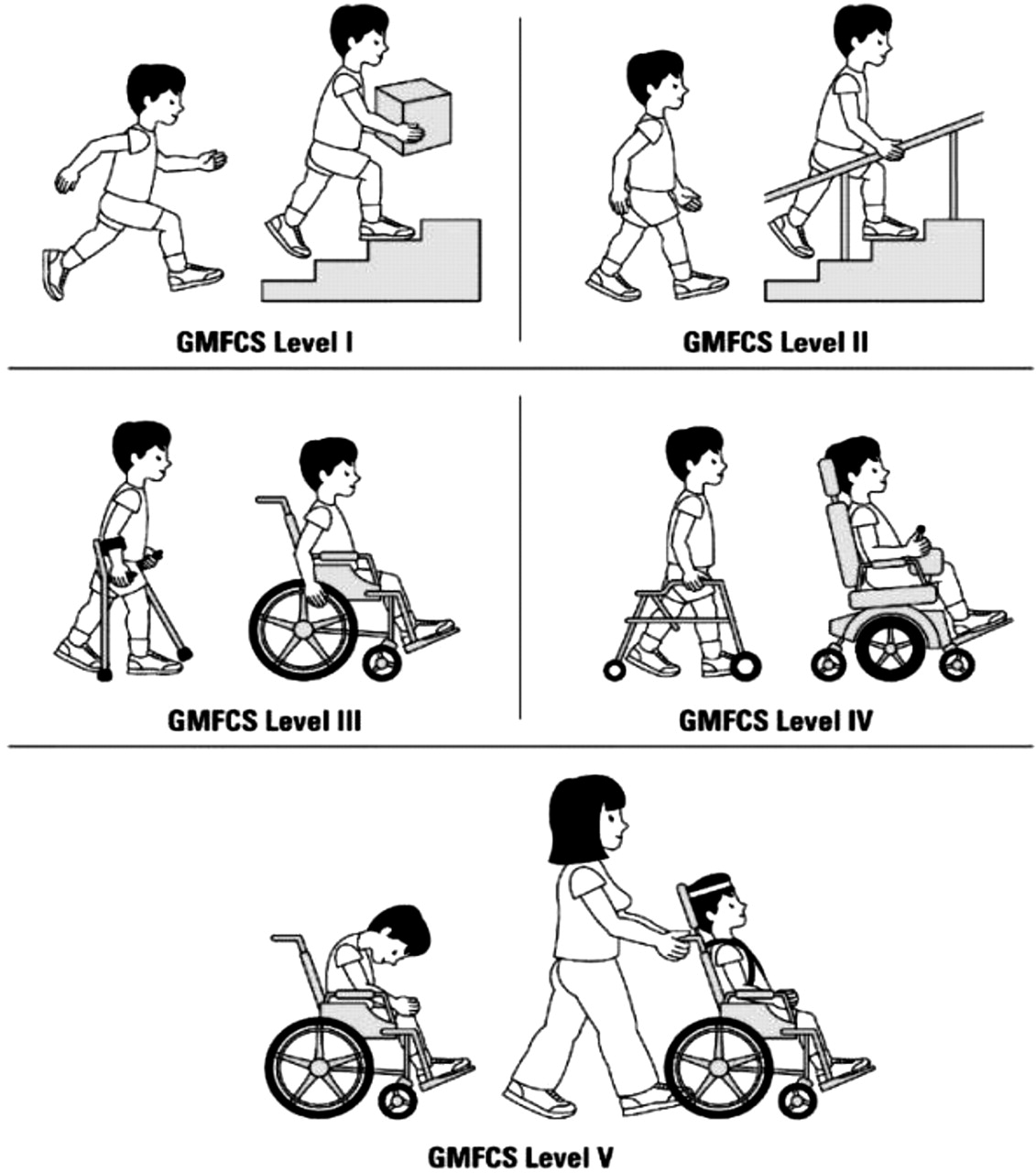
▶ The gross motor functional classification system (GMFCS – figure 1) is used clinically to delineate individuals with CP into five groups dependent on their level of mobility; – level one being completely independent and five totally dependent.5
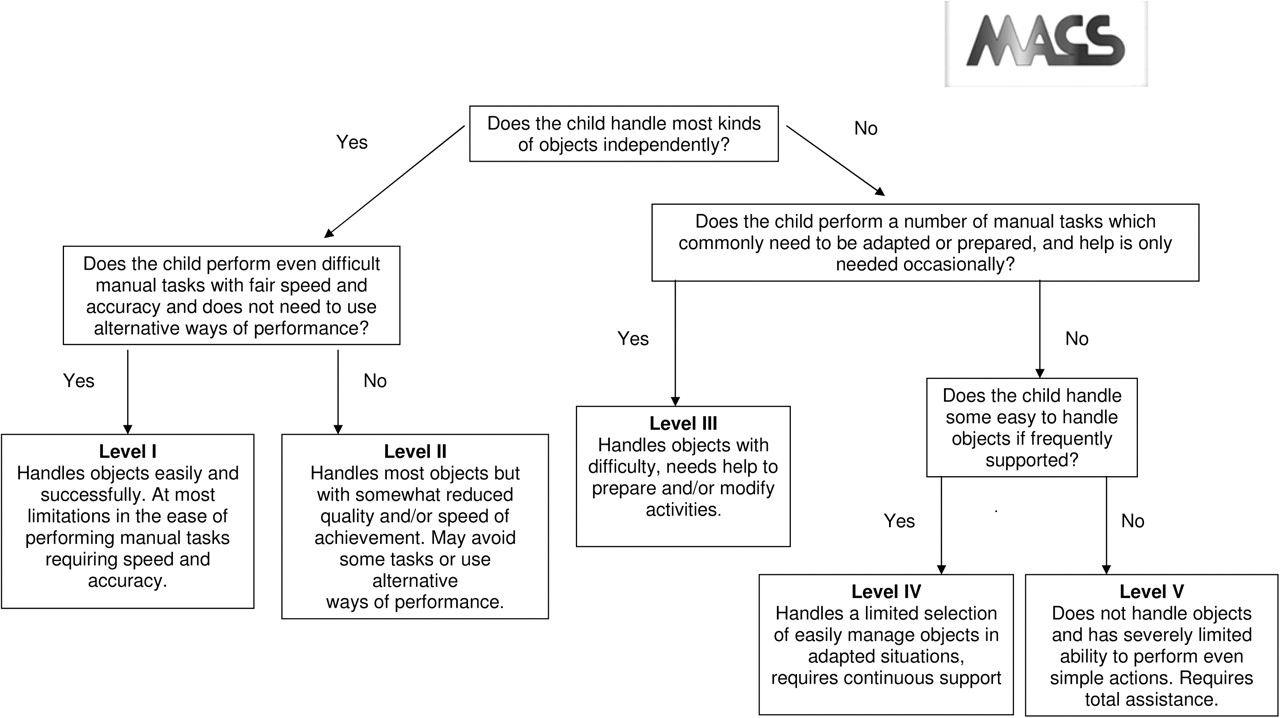
▶ The Manual Ability Classification System (MACS – figure 2) does the same for arm and hand function.6
▶ As yet the oro-motor system and swallowing has not been looked at in such detail, but its dysfunctional effect on activity and quality of life can be just as marked as the other more ‘obvious’ motor difficulties.

The gross motor functional classification system (GMFCS).

The Manual Ability Classification System (MACS).
The motor pattern of secondary abnormal muscle tone observed is almost universally associated with the site of the underlying primary pathophysiology, though, as all clinical problems are individual, total reliance of neuroimaging for diagnosis is not entirely reliable.
▶ Spasticity – velocity dependent increase in tonic stretch reflexes causing hypertonia, which is associated with the ‘positive’ side of the upper motor syndrome. Simplistically, the descending cortico-spinal tracts normally stimulate release of the inhibitory neurotransmitter γ-amino butyric acid (GABA) at the spinal level. Lesions lead to dysinhibition of the spinal reflex arc and muscle over activation. Treatment policies focus on muscle rigidity/hypertonia rather than the spasticity itself. While we focus on the corticospinal tracts, there is considerable neuro-physiology research on the interplay with other pathways of descending motor control.3 ,7
▶ Functionally, the ‘negative’ symptoms of the upper motor syndrome – reduced motor activity leading to weakness and poor selective motor control are far more disabling for the individual and more difficult for us to treat.
▶ Dystonia – On the other hand, damage primarily to the basal ganglia leads to patterns of sustained disturbed muscle contraction causing abnormal postures that are frequently associated with involuntary movements especially with sensory ‘overload’ such as anxiety, noise or discomfort – dystonic CP. These strange, non-sustained movements can of course be further subdivided, for example, chorea and athetosis, but the overriding term of dystonia, helps focus treatment. The term does not include rare ataxic forms of CP, which generally arise from impairment of the cerebellum. The term dyskinesia is often used in a similar context – dys+kinesis=abnormal+movement, so fluctuating movement pattern.
▶ It is of course more common to see a combination of dystonic and ‘spastic’ elements rather than true dystonia within an individual's movement disorder.
Looking at the prevalence of different motor patterns, all have remained remarkably static over the last 20 years, though the complexity may have increased with neonatal survival.1
▶ 1.2–1.5 per 1000 – bilateral spastic CP;
▶ 0.6–0.8 per 1000 – unilateral or hemiplegic CP and
▶ 0.15–0.25 per 1000 – bilateral dystonic CP.
Differential diagnosis
When the clinical team assesses any child with a movement disorder, the first question should be what is the cause? Once again, it is important to stress that CP is not a diagnostic but a descriptive term. It is vital to ensure that a progressive or rarely treatable form of a movement disorder is not missed. Clear early obstetric and neonatal history and neurological examination are of greatest aetiological importance. However, even if an ante, peri or postnatal cause is given, if the pattern of on-going development does not continue as expected, or if other body systems are involved, further investigation may be necessary to look for a potential cause, including neuro-imaging and appropriate assays. Indeed the recommendations of the American Academy of Neurology and SCPE are that all children with suspected CP of uncertain origin should have cerebral imaging.1 ,8
Differential diagnoses include genetic, metabolic, mitochondrial and progressive neurological and neuro-muscular conditions. There are numerous rare differentials but consideration of a dopamine or partial dopamine responsive disorder is particularly important in dystonia, when a short therapeutic trial of L-dihydroxyphenylalanine (DOPA)can make a phenomenal functional difference. If there is any question of aetiology, referral to a specialist regional centre is appropriate. However, routine screening metabolic tests alone are unlikely to provide further information.9
Interventions
Presentation is generally either during neonatal follow-up with an ‘at risk history’ or from primary care screening for neurodevelopmental delay. Often, abnormal muscle tone is obvious from an early stage or else comorbidities such as epilepsy, gastro-oesophageal reflux and feeding difficulty are clinically overwhelming. If present, dystonia develops only when the neural pathways are sufficiently mature – around 18 months of age; prior to this the child is usually extremely irritable with a background of low or high tone.
Overview of multidisciplinary management
Appropriate management of children with CP is possible only with a multi/interdisciplinary team approach, keeping the child and family at the centre of all decision making processes, as always, the parent is the world expert on their child.
Paediatricians depend on the expertise of numerous colleagues, including neuro, general and orthopaedic surgeons, gait analysis laboratory teams, physio-, occupational, speech and language and psychotherapists, orthotists and nursing specialists, charities, teachers and social workers, all working together to reduce the functional effect of the physical impairment on the child.
Initial evaluation focuses on what is important for the child and family in terms of the following:
▶ movement, posture and mobility,
▶ communication,
▶ comorbidities,
▶ activity of daily living,
▶ care,
▶ comfort and
▶ quality of life.
Regular re-assessment is necessary to look at neurodevelopment and the outcome of interventions. As discussed above, these obviously vary with the severity, distribution and type of CP encountered. In particular, recognition and management of pain can be very difficult in children with severe CP who may have limited communication and multiple clinical difficulties.
Multidisciplinary motor management
The principle is to focus on the functional impact that the disability has on the whole individual. In paediatrics, we need to respond to the dynamic processes of skeletal and soft tissue growth, to minimise secondary-biomechanical deformities. We provide an adaptive approach preparing the child for adult mobility, using different treatment modalities, working with, rather than against background movement and posture.
The core programme for any child is co-ordinated therapy, to optimise potential and minimise disability. There are a multitude of different physical and occupational therapeutic modalities.10 Individual child development centres will have slightly different approaches, but most are based on the principles of neural plasticity, patterning, postural balance, muscle strengthening and stretching. Adaptive approaches to remodelling or ‘Bobath’ therapy neurodevelopmental training are used in techniques such as ‘Vojta’ therapy or conductive education, as championed by the Peto institute, which are used widely across the UK.11
Orthotics (the use of rigid polypropylene, lycra or neoprene splints) and postural equipment are used widely when appropriate, to facilitate symmetrical sitting, standing, lying, mobility and toileting. Speech, communication and oro-motor control are vital to the developmental progression of any child; however, we will here focus primarily on medical movement therapies.
Specific medical movement therapies
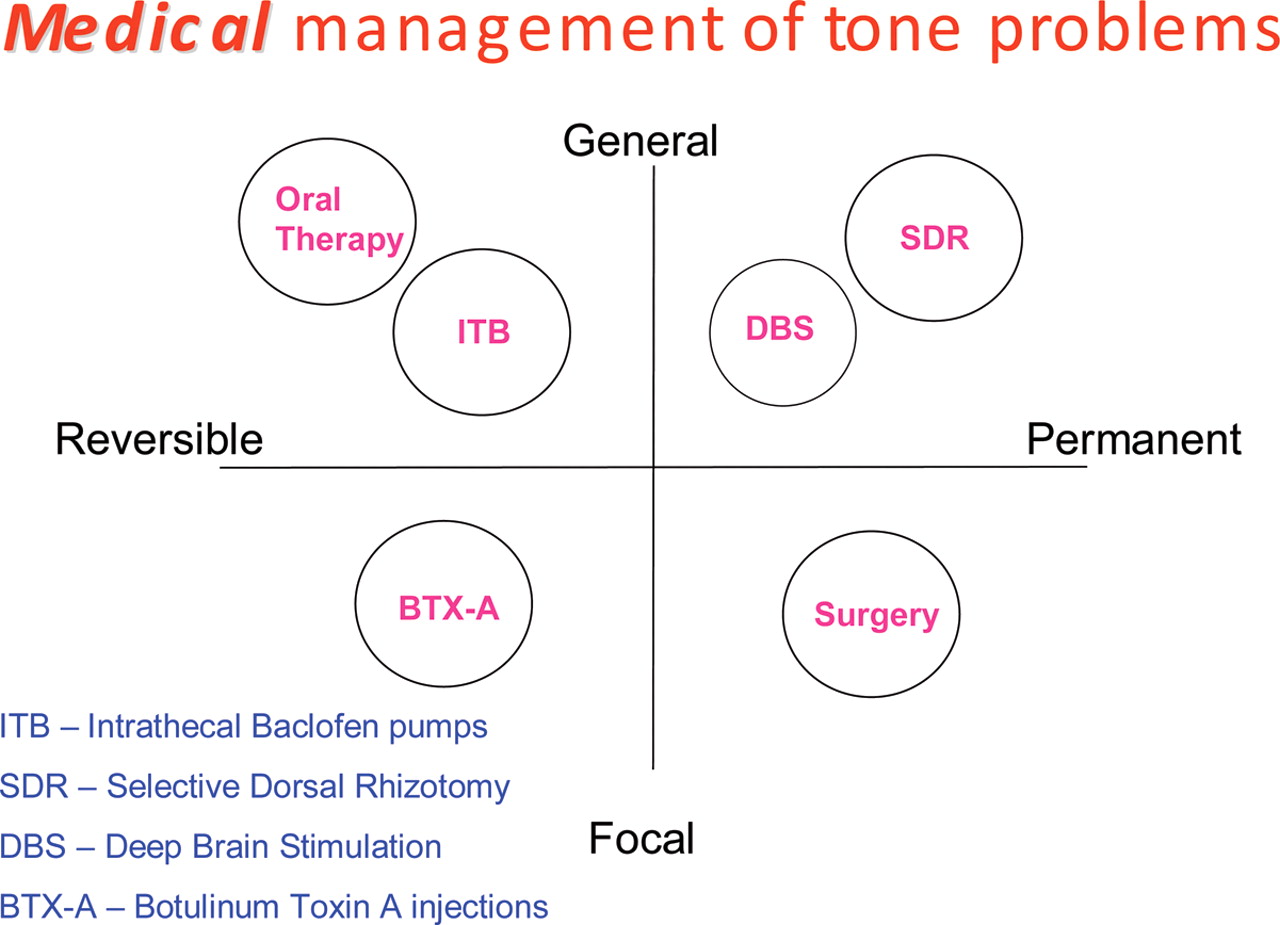
Specific medical movement therapies, focus on the child as a whole or part (ie, limb) and treat in a reversible (ish) or permanent (ish) fashion (figure 3). Two words of caution:
▶ first, we are managing the secondary problems of the movement disorder and not dealing with the primary impairment.
▶ second, it is important to stress that they all work as adjunct to rather than taking the place of day-to-day therapy programmes.

{kind=link}
{kind=link}
{kind=link}
Medical movement therapy options.
Medically, we focus on the muscle rigidity/spasticity, dystonic movements and comorbidities seen in young people with CP.
Medical movement therapies
Oral medicines
Anti-‘spasticity’ agents
Several unlicensed medicines are used commonly to directly or indirectly influence high-muscle tone rather than to treat spasticity per se. They are outlined in figure 4 and table 1.12 Anticonvulsants, such as carbamazepine and levetiracetam also have movement-modifying effects, with potential efficacy in movement disorders alone.
Some of the more commonly used oral medicines
Notes of caution:
▶ all have significant side effects and limited evidence base beyond personal practice.
▶ often in severe CP, there is a combination of limb hypertonia in conjunction with truncal hypotonia. The latter is exacerbated by muscle relaxants and can be equally, if not more, disabling.
▶ Individuals can use their increased tone to improve function in, for example, standing or transfers. Reduction in tone, therefore, has an effect on the daily activities.
Antidystonic medication
If there is a significant element of dystonia observed, then medicines for Parkinson's or Huntington's disease are increasingly used in children with CP. The postulate is to optimise function of the damaged basal ganglia by increasing the amount of neurotransmitter available.13 ,14
Subtle but significant improvements in the fluidity of motor control can be gained; those seen include better oro-motor control and truncal balance, reduced extensor spasms and easier hand function. Initiation and supervision of such treatment should be in specialist units.
An understanding of the management of status dystonicus is necessary as this is a real and profound medical emergency. There is no overriding consensus, but generally intravenous clonidine or high-dose benzodiazepines are used followed by artificial initiation of sleep, as this abolishes all dystonic movements.15
Invasive options
Botulinum toxin A injections
Botulinum toxin A (BTX-A) was first used to reduce focal spasticity in CP in the mid-1990s. It is licensed only for use ‘in children over the age of 2 for dynamic equinus foot deformity caused by spasticity in ambulant paediatric CP.’ However, there has been considerable publication on its use in other muscles and glands that cause focal, functional difficulty, including multilevel lower and upper limb, hip (for pain relief), neck and paraspinal muscles as well as salivary gland injections. There is good level 1 evidence for its use at calf, hip adductor, hamstring and upper limb levels. The American Academy of Neurology has stated that it should be universally offered as a treatment option during childhood.12 ,16
Alternative treatments used internationally for the management of CP
Evidence-based guidelines are regularly updated with the most recent being the 2009 European and 2010 International consensus statements.17 These focus on medicolegal and medicoeconomic factors, common indications and integration with other therapy modalities; appropriate assessment, accepted off license use, dosage, safety, administration, re-evaluation, continuation and discontinuation.
Three products are commercially available: Botox (Allergan, Irvine, California), Dysport (Ipsen, Paris, France) and Xeomin (Merz, Frankfurt, Germany). Their dose ranges are not interchangeable, with individual different pharmacokinetic and pharmacodynamic responses.
This potent neurotoxin is taken up in neuromuscular junction end plates due to its high affinity for the synaptic vesicle protein 2 which is upregulated in active neurons. The cleaved short chain then targets the SNAP-25 protein of the acetylcholine release complex, thereby irreversibly blocking the motor end plate potential. Synaptic regression then occurs and a new bud resprouts from the end of the axonal sheath. This is why the effect of focal muscle relaxation lasts for around 3 to 4 months. Clinical benefit often lasts for a longer period of time due to the prolonged muscle relaxation and stretch. Weakness is exacerbated and there are increasing concerns about long-term effects if used too frequently.18
Multidisciplinary assessment of muscle function and dysfunction and correct administration to targeted areas is vital. The use of electromyography (EMG), muscle stimulators or direct ultrasound guidance for placement is increasingly accepted as the best practice. It is not a treatment to be used without consideration of postinjection guidelines, such as those provided by the Association of Paediatric Chartered Physiotherapists.19
Intrathecal baclofen pumps (ITB)
These have been used in paediatrics for over two decades. The aim is to maximise antispasticity benefits of baclofen at the spinal GABA-ergic level and minimise the cerebral side effects, by using an implantable pump. Oral baclofen does not cross the blood–brain barrier easily as it is poorly fat soluble.
A recent 2010 European Consensus on the appropriate use of ITB therapy in paediatric spasticity has been published.20 This focuses on its use as an ‘antispasticity measure’, though it is also of benefit when both muscle hypertonia and dystonia are present.
The evidence is strong for use in young people with a severe motor disability – GMFCS levels 4 and 5, though there is less published work on its use in children who are independent or therapeutic community walkers – GMFCS levels 1, 2 and 3, the population group increasingly targeted. An appropriately used ITB can improve care, activities of daily living and quality of life.
A trial of oral medication is initially given to deduce the balance of effect and side effect. As always, careful assessment and clear understanding of the procedure and the possible outcomes, by all the members of the family is necessary.
A screening trial of 50 mcg is given intrathecally, as a bolus, to observe any functional improvement. If successful, the neurosurgical teams insert the pump and catheter. Standard rates vary individually but are usually in the range of 0.05–1.25 mg per day. Refilling needs to occur every few months, through an internal portal; syringe and needle, doses can be regulated at that time.
Overdose tends to lead to hypotonia, sleepiness and respiratory depression. Clinical signs of withdrawal include increased muscle tone, agitation, hallucinations, itching, hyperthermia, seizures and unless recognised in time multi organ failure and muscle rhabdomyolysis.
Selective dorsal rhizotomy
Surgical resection of nerve rootlets in the dorsal spinal root can reduce the stimulation of the spinal reflex arc, a procedure first carried out in the early 1900s. Exceptionally careful physical, functional and electrophysiological assessment is necessary, to minimise risk and optimise potential, as any such intervention is irreversible.
A minimal laminectomy is performed, the motor and sensory nerves are separated and then the sensory fibres are blunt dissected into a series of rootlets. On table EMG stimulation elicits peripheral motor effect. A proportion of rootlets are then cut between 25 and 50% of L2 to S2.
Recent National Institute for Health and Clinical Excellence guidelines for the use of SDR in paediatric spasticity have been collated.21 Published effects are variable and though short-term benefits can be marked, long-term functional difference is less reproducible.22 Evidence of benefit is seen primarily in reducing moderate to severe lower limb spasticity – GMFCS levels 4 and 5. It is also increasingly being used in the independent walking population – GMFCS levels 2 and 3. Great care must be taken not to critically weaken the muscles at the expense of reducing rigidity. In published series, up to 30% of individuals have irreversible side effects such as back pain, increased scoliosis, sensory loss, weakness, neuropathic bowel and bladder.
The skill of the whole team is therefore vital for outcome, with a prolonged period of focused rehabilitation absolutely necessary. Although assessment and intervention is offered in several regional centres, there is a current trend with children accepted onto expensive intervention programmes from units outside the UK. With long distance and limited appraisal, there are potential dangers to the child, raised expectations for the families and the burden of unplanned expensive rehabilitation for NHS services.
In comparative papers, the invasive options of BTX-A, ITB, SDR and multilevel orthopaedic surgery seem to have equal mid- to long-term outcomes.23,–,25
Deep brain stimulation
In young people with severe dystonic quadriplegic CP, implanting quadripolar electrodes within the basal ganglia of the brain can deliver a continuous electrical signal to the target nuclei. The globus pallidus interna is usually targeted and by doing this aberrant signals from the damaged locomotor driving system are felt to be made more organised.26
The insertion of this ‘brain pace maker’ has led to the improvement in dystonia rating scales between 5% and 40%, as well as the general quality of life. An intact corticospinal tract is necessary for benefit so any extensive damage of this is a contraindication. In this case, insertion of an intrathecal baclofen pump would be considered instead.
Obviously assessment, implantation and on-going management should only occur in specialist centres. Side effects are not uncommon and generally it is only considered if all other avenues of treatment have not helped provide a comfortable quality of life. There is at present little published work looking at the long-term benefit in children with secondary movement disorders such as CP but short-term improvement in quality of life is also significant.
Orthopaedic surgery
Orthopaedic screening in bilateral CP is vital. Early hip screening and intervention programmes can make a dramatic effect on comfort, care and quality of life by reducing subluxation and dislocation.27 Later, spinal curvature needs regular assessment. Soft tissue and bony surgical management is often core to the holistic well-being of the child. Timing of any intervention is vital, obviously a child should be better off in the long term having had surgery than they would have been without it.
Generally surgical options are
▶ to lengthen shortened muscle–tendon complexes,
▶ move awkward muscles,
▶ bio-mechanically improve lever arms or
▶ provide a stable base for standing and/or walking.
Mobility, particularly maintenance of independent transfers, comfort and care are all specific functional goals to work towards. In the walking child, as with botulinum toxin A injections, multilevel intervention with early rehabilitation is now considered best practice, rather than piecemeal annual surgery at the ankle, knee and hip.28 The gold standard assessment for any such surgery is the use of the formal gait analysis comprising the 3-D kinematic and kinetic evaluations.
Musculoskeletal scoliosis is a major risk, especially in children with severe CP. Specialist intervention via insertion of spinal rods or vertebral fusion can reduce spinal curvature and improve the clinical state, quality of life and ease of care for individuals. Morbidity rates are high, particularly in children with a background of poor nutrition, uncontrolled muscle spasms and compromised clinical state.
Stem cell therapy
The potential for treatment with pluripotential stem cells is felt by some to be the nirvana of management options.29 We are not just minimising the impact of secondary deformity, but are able to focus on the primary impairment itself. Early intervention, when the brain cells and tracts have maximum potential for plasticity would be ideal.
Whether by using intravenous, intrathecal or direct intracerebral injection routes, if you read the internet alone (Google=2 320 000 hits; cheap at $30 000.00) we are already there. Unfortunately, as yet, there is no reliable medical evidence of short, mid or long-term benefit in spite of numerous impressive individual case reports from centres in Germany, China, India and the Middle East. These should be balanced by the side effects seen in epilepsy, infection and worsening of neurological dysfunction that have been reported, particularly in techniques implanting directly into the central nervous system.
Though the basic science of regenerative neurology is rapidly expanding and practice is becoming increasingly standardised, there is certainly no possibility of its widespread acceptance as mainstream medical practice, at present. However, watch this space….
Complementary medicine
There are a myriad of other forms of complementary or alternative treatments used internationally for the management of CP.30 ,31 Many have been found to be very beneficial in individual cases.
For most complimentary treatments, there is generally a poor evidence base, with very few randomised controlled trials or even case series showing long-term benefits. In the absence of definite evidence-based practice and where there is no obvious ‘cure’, un-proven alternative options are often an attractive option. Without careful regulation there is a tendency for malpractice and considerable expense to the family. The support of the traditional medical establishment in setting up clinical trials and regulatory bodies is vital. Without these, as open-minded advocates for our clients, we have to remain open, yet guarded to the claims of such therapeutic options.
Management of comorbidities
It is vital to stress that the management of comorbidities is the key for children with CP. Dependent once again on the severity and type of underlying movement disorder, the prevalence of other clinical problems can be extremely high.
Just from a gastrointestinal viewpoint, these include specific and general nutrition and growth problems (including poor wound healing and osteoporosis), oro-motor difficulties (including uncoordinated swallowing and poor saliva control), upper and lower tract dysmotility, gastro-oesophageal reflux, malabsorption and constipation (including mega-colon). High rates of immune difficulties, chest, urinary tract and skin infections, intractable epilepsy, pain from a myriad of sources including osteoarthritis; sleep, sensory, including visuoperceptive problems, developmental and behavioural difficulties are seen. Obviously, the holistic skills of a general paediatrician are vital!
The adult with CP
There is an increasing acknowledgement by rehabilitation colleagues of the specific challenges in managing the adult population group with CP. A key part of the duty of individual paediatricians is to ensure a smooth transition in the patient pathway. An excellent review on the specific clinical challenges was provided in a special supplement in Developmental Medicine and Child Neurology in 2009.32
Conclusions
Any care pathway for children with a physical neurological impairment depends on a child- and family-centred approach, with the need for careful communication between all the myriad health, education and social services that are likely to become involved. Holistic care depends on early recognition and intervention in the developmental, physical and psychological difficulties that such children regularly encounter.
With the high prevalence of children with non-progressive motor disorders, it is important that all child health workers have an understanding of the basic mechanisms of neurological, biomechanical and pharmacological management of children with CP and how that may have an effect on their general or specialist areas of intervention. The care of children with CP relies on all paediatricians, not just those who have developed a specialist interest in the area.
More specifically, the variety of potential medical movement therapies has increased greatly over the past few decades. With the injudicious use of the internet, families desperate to seek miracle cures are rightly demanding specialist assessment and intervention but the evidence base for many procedures is patchy at best. Randomised control trials with a baseline of no intervention are difficult to countenance in a situation where to do nothing puts the child at risk of developing secondary deformity. However, over the next few years both in the UK and in Europe the aim of specialist working parties will be to provide clinicians with careful guidance of what motor management policy is appropriate, at what age and developmental level.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.