Article Text

Statistics from Altmetric.com
Introduction
Streptococcus pneumoniae, the pneumococcus, is a Gram-positive diplococcus with more than 90 serotypes determined by the polysaccharide composition of its capsule. It is carried asymptomatically in approximately 50% of people.1 It can cause both non-invasive and invasive disease. Invasive pneumococcal disease (IPD) refers to disease in which the bacterium enters a sterile site such as blood, cerebrospinal fluid (CSF), pleural fluid, joint fluid or pericardial fluid. Non-invasive disease includes otitis media, sinusitis and bronchitis. Among infectious diseases, IPD is a leading cause of morbidity and mortality in children and adults. Vaccination against pneumococcal infection was introduced into the UK routine childhood immunisation programme in 2006 with Prevenar 7 (Pfizer, Tadworth, UK), a seven-valent conjugate vaccine, replaced in 2010 with the 13-valent vaccine Prevenar 13. Pneumococcal disease continues to be a problem despite these measures.
Invasive pneumococcal disease
In order to make a diagnosis of IPD, pneumococcus must be isolated from a sterile site, either by culture or by detection of bacterial cell surface antigens. In the UK, all isolates from IPD, irrespective of patient age, should be referred for serotyping to the Health Protection Agency (HPA) pneumococcal reference laboratory.
Children with pneumococcal infection often present with non-specific symptoms and signs including isolated fever. National Institute for Health and Clinical Excellence (NICE) guidance for management of feverish illness in children should be followed, including administration of antibiotics which cover IPD when serious bacterial infection is likely or suspected.2
Certain populations of children are at increased risk of IPD. Children at particularly high risk are those with antibody deficiencies, such as X linked (Bruton's) agammaglobulinaemia, anatomic or functional asplenia, sickle cell disease, HIV infection and those with cochlear implants or CSF leak. Children with other chronic illness (eg, cardiac, pulmonary or renal disease and diabetes mellitus) and those receiving chemotherapy or steroids are also at risk. Children in day care from an early age also have a higher incidence of IPD.3 4 Around 10–20% of children with pneumococcal meningitis have an underlying comorbidity.5 6
Many children with IPD present severely unwell as a consequence of pneumonia, septicaemia or meningitis and require intensive care. The assessment and management of these children should follow the standard ABCapproach, with appropriate supportive therapies for shock or neurological failure including intubation, ventilation, volume resuscitation and inotropic support as necessary. It is not within the scope of this article to discuss supportive measures for septic shock in further detail. However, in such cases, early referral to the regional paediatric intensive care unit (PICU) for advice on management and retrieval is recommended. Once an initial assessment of severity has been made and supportive measures instituted, appropriate investigations should be arranged to identify the site of infection so that specific therapy may be commenced.
Pneumococcal meningitis
Meningitis is inflammation of the meninges covering the brain and spinal cord. Pneumococcal meningitis usually develops after pneumococcal bacteraemia, present at a sustained high level for 12–24 h before bacteria cross the blood–brain barrier. The bacteria multiply on entry to the CSF, releasing components of the cell wall and the bacterial enzyme pneumolysin. These stimulate host cell release of cytokines, increasing inflammation and vascular permeability and stimulating infiltration of leucocytes. Changes in the CSF occur within hours of infection.
S pneumoniae is the second most common cause of bacterial meningitis in children over the age of 1 month in the UK after meningococcus.7 In younger children, pneumococcal meningitis often presents with non-specific features including fever, irritability, difficulty feeding and vomiting. These are also common features of viral respiratory tract infections, which often precede blood stream infection with pneumococcus as they facilitate pneumococcal colonisation and invasion. This can make early diagnosis difficult. As the disease progresses, focal neurology, seizures and reduced conscious level commonly develop. Indeed, pneumococcal meningitis is more likely to present with focal neurology, coma (11%) or seizures (20%) than other forms of postneonatal bacterial meningitis.8 Older children more commonly present with classic features of meningitis such as headache, fever, neck stiffness and altered mental state. The main differential diagnoses include other infective causes of meningoencephalitis, cerebral abscess or other mass lesion.
Although examination of CSF obtained by lumbar puncture is diagnostic, lumbar puncture is often contraindicated in the acute setting where there may be symptoms or signs of raised intracranial pressure (table 1)9 and therefore a risk of cerebral herniation following lumbar puncture.10 In a patient in whom lumbar puncture is deferred, antibiotic therapy should be commenced immediately following collection of blood for culture, rapid antigen testing and PCR. Confirmation of pneumococcal meningitis can still be made following deferred lumbar puncture with antigen detection or PCR. CT scanning is helpful to rule out other causes of focal neurology, altered mental status or seizures. However a normal CT scan does not guarantee absence of risk from lumbar puncture.11 Other recommended laboratory investigations are shown in table 2.
Contraindications for lumbar puncture in suspected acute bacterial meningitis9
Recommended laboratory investigations in acute bacterial meningitis9
Treatment should include specific antibiotic therapy targeted to all causes of bacterial meningitis, in addition to general supportive care including intensive care if necessary. Antibiotic therapy should be initiated as soon as possible. In the prehospital environment antibiotic treatment (usually benzylpenicillin intramuscularly or intravenously) should be commenced if there is a strong suspicion of meningococcal disease or if an unacceptable delay (>90 min) is anticipated before the patient arrives in hospital. In the UK hospital environment, third-generation cephalosporins which cross the blood–brain barrier, such as cefotaxime or ceftriaxone, are recommended as first-line antibiotic therapy. In patients with a known history of β-lactam allergy, vancomycin should be administered as the alternative in pneumococcal meningitis. In regions such as Spain and the USA where penicillin-resistant strains of pneumococcus are common, vancomycin should be administered in combination with a third-generation cephalosporin. Once culture sensitivities are available, antibiotics can be changed if necessary. Antibiotic therapy should be continued for 10–14 days depending on clinical response and decrease in markers of inflammation.9
Use of steroids is controversial. NICE has published guidelines on bacterial meningitis, including steroid use in September 2010.12 NICE recommends dexamethasone if there is suspicion or confirmed bacterial meningitis in a child over 3 months old, at a dose of 0.15 mg/kg (to a maximum of 10 mg) 6 h for 4 days. Dexamethasone should be started as soon as possible, before or within 4 h of antibiotics but certainly no more than 12 h after initiation of antibiotics. This concurs with current guidance from the American Academy of Pediatrics Committee on Infectious Disease. A recent Dutch study in adults also suggests that use of dexamethasone may reduce morbidity and mortality.13
Complications of pneumococcal meningitis include seizures, increased intracranial pressure, cerebral oedema, focal cerebral ischaemia14 (figure 1) and infective subdural effusions. Some cases of pneumococcal meningitis develop rapidly progressive cerebral oedema that may result in raised intracranial pressure, generalised ischaemia and death (figure 2). If signs of raised intracranial pressure are present, general neuroprotective measures to maintain a good supply of oxygenated haemoglobin to the brain may be helpful if the child is in intensive care. These include maintaining a 30° head up tilt, haemoglobin >10 g/dl, ensuring a good blood pressure and maintenance of arterial carbon dioxide tension within the normal range. Seizures should be treated with a parenteral anticonvulsant, such as phenytoin. There is currently no evidence to support the routine use of intracranial pressure monitoring or surgical measures in non-traumatic cerebral oedema.9

CT brain scan in pneumococcal meningoencephalitis, showing focal infarction in the right frontal lobe and the left frontal and parietal lobes. This pattern of infarction is due to focal vascular occlusion rather than generalised swelling.
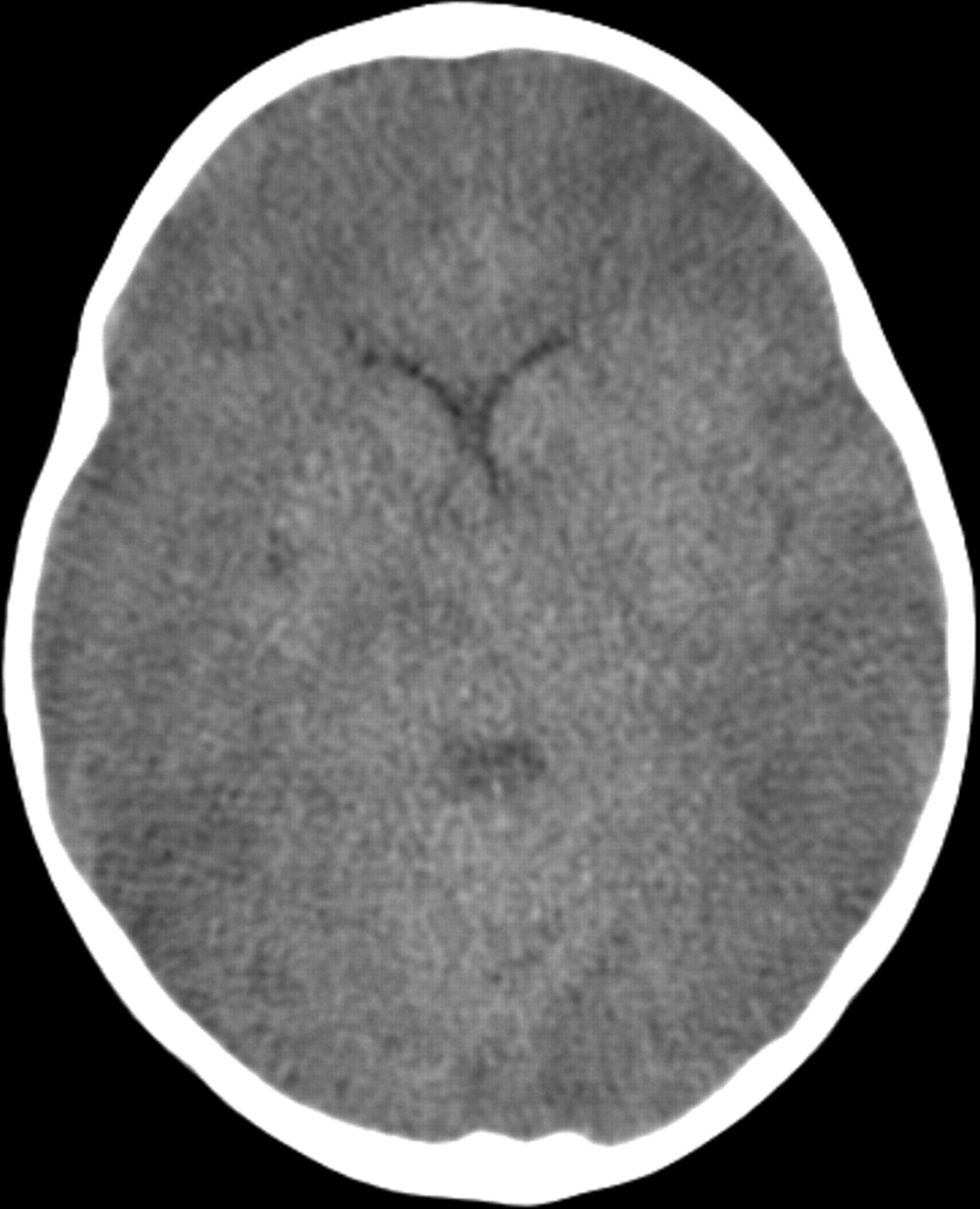
CT brain scan in pneumococcal meningoencephalitis, showing widespread areas of infarction due to generalised brain swelling.
In pneumococcal meningitis, there is a high incidence of serious neurological consequences in survivors, including cognitive and motor deficits (25%) and hearing loss (32%).8 Survivors of pneumococcal meningitis require hearing tests and some need long-term neurodevelopmental follow-up. Mortality is higher in pneumococcal meningitis than in meningococcal or Haemophilus influenzae meningitis, at approximately 8% for children in developed countries.5 8 There is a higher risk of death if children present in shock or in a coma or require mechanical ventilation.8
Pneumococcal pneumonia
Pneumococcal pneumonia follows aerosolisation of bacteria from the nasopharynx to the alveoli. The pneumococci multiply in the alveolus, invade the alveolar epithelium and pass through the pores of Cohn to adjacent alveoli, causing inflammation and consolidation in lobar pattern. Over 2 or 3 days an inflammatory exudate accumulates, adding to local damage. Following this, pneumococci are phagocytosed by leucocytes, fever ceases and exudates slowly resorb, usually with minimal scarring.
Pneumococcal pneumonia is the most common cause of community-acquired bacterial pneumonia in children under 2 in the UK15 and often occurs in combination with a concurrent viral infection of the upper respiratory tract. Presentation is often as a lobar bacterial pneumonia (figure 3), the most common symptoms being fever, cough and tachypnoea. The diagnosis may be made clinically, on chest radiography and from culture, PCR or antigen testing of blood or antigen testing of urine. As only 10–15% of children have positive blood cultures if hospitalised for pneumonia, real time PCR for pneumococcal DNA from clinical samples is becoming increasingly useful. If pleural fluid is sampled, testing for pneumococcal antigen or DNA may give the diagnosis even if antibiotics have already been given.16
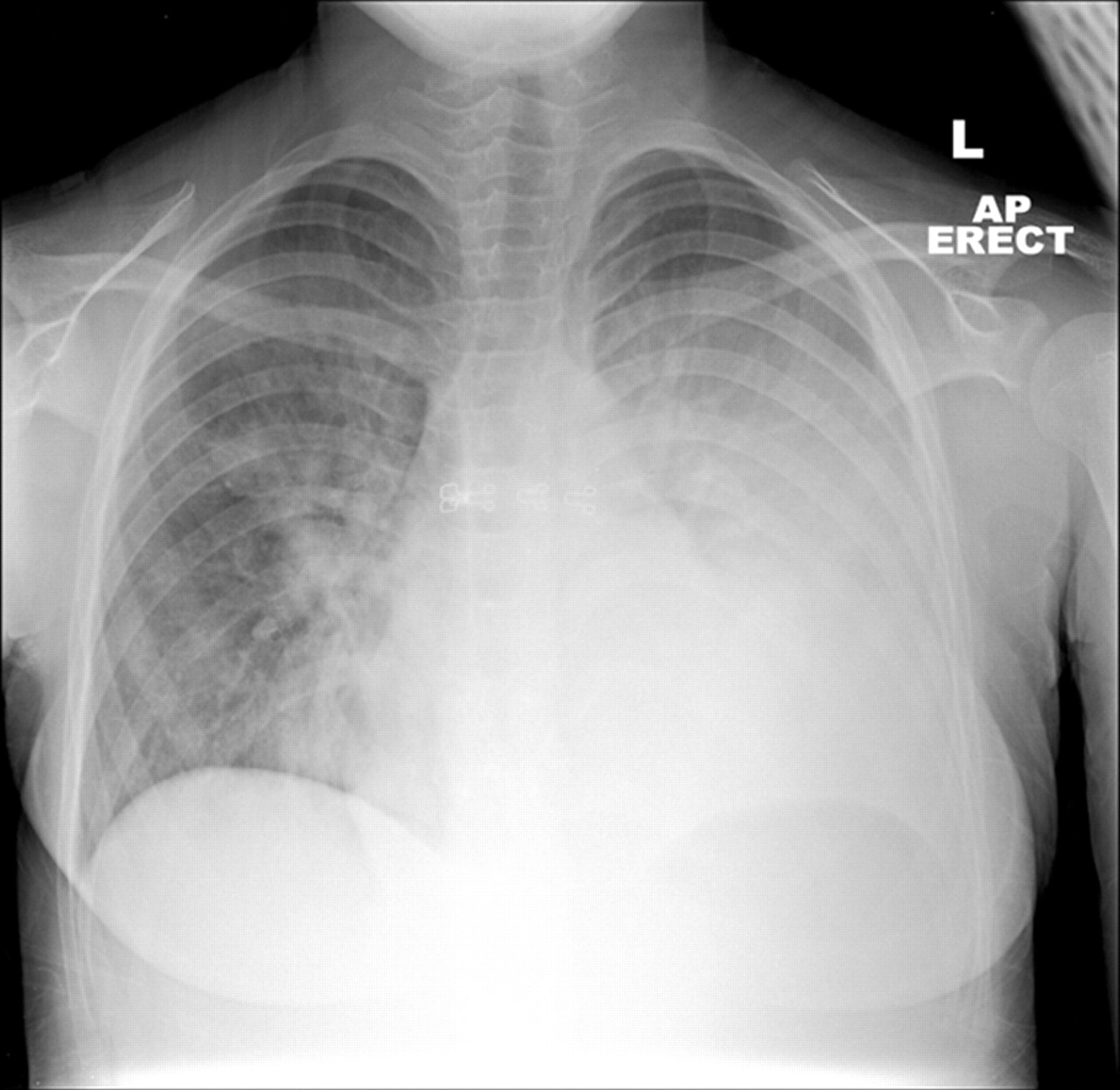
Chest radiograph in pneumococcal pneumonia, showing left lower lobe consolidation.
Antibiotic treatment is usually empirical, as in most cases of bacterial pneumonia, isolation of an organism is the exception rather than the rule. Oral antibiotics may be suitable for children who are systemically well. If intravenous antibiotics are necessary, the first line in the UK is a third-generation cephalosporin such as cefotaxime or ceftriaxone, which also cover other common causes of community-acquired bacterial pneumonia. If cephalosporin resistance is considered likely, or in the presence of very severe infection or empyema, vancomycin or clindamycin may be added. Treatment is usually for 5–7 days or until the child is afebrile for 5 days in severe cases.
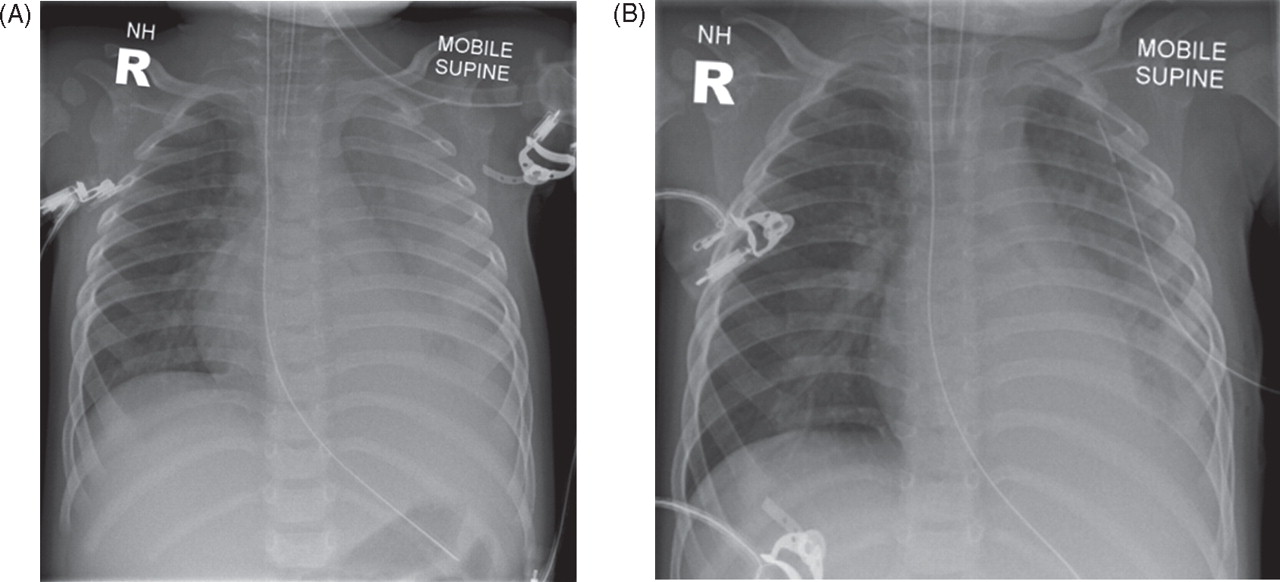
Pneumococcal pneumonia is often uncomplicated, with complete recovery. A significant and common complication of pneumococcal pneumonia is pleural effusion and empyema (figure 4A,B), which continue to be seen despite vaccination.17 Drainage can be with either pig tail drains or ordinary chest drains; surgical decortication may be necessary in the management of complicated empyema. Intrapleural instillation of urokinase may be of benefit aiding drainage of empyema.18 Patients should be managed according to the guidelines from the British Thoracic Society.19 Advice from cardiothoracic and respiratory specialists may be helpful in managing these children. Other possible complications of pneumococcal pneumonia include lung abscess and necrotising pneumonia. A small proportion of children develop very severe respiratory failure associated with widespread consolidation, resulting in acute lung injury (ALI) or acute respiratory distress syndrome (ARDS), the severe end of the spectrum of ALI (figure 5). The diagnostic criteria for these conditions were developed in 1994 by an American–European Consensus Conference20 (table 3).

Chest radiographs showing left-sided parapneumonic effusion due to pneumococcal pneumonia, (A) before and (B) after drainage.
Chest radiograph showing four-quadrant airspace shadowing in acute respiratory distress syndrome due to pneumococcal pneumonia and septicaemia.
Diagnostic criteria for ALI and ARDS
ALI is characterised by widespread airway collapse, surfactant deficiency and reduced lung compliance. It is an inflammatory disorder with three phases: exudative, proliferative and fibrotic. The exudative phase lasts for the first week and is marked by alveolar oedema with formation of hyaline membranes. In the proliferative phase, in the second week, organisation of the inflammatory exudate occurs. In the fibrotic phase, which may occur from day 10, pulmonary fibrosis is seen. However, many studies show that survivors return to normal lung function, with complete resolution of pulmonary fibrosis.21
Patients with ALI or ARDS usually require mechanical ventilation. The goals of treating patients with ALI/ARDS are to maintain adequate gas exchange while avoiding ventilator-induced lung injury, to treat the underlying cause of the condition and to attenuate the inflammatory response. High-concentration inspired oxygen should be avoided to limit the risk of direct cellular toxicity and to avoid reabsorption atelectasis. Arterial oxygen saturation values of around >88% or lower are commonly accepted. A ‘lung protective ventilation strategy’, optimising positive end expiratory pressure, using a tidal volume of <6 ml/kg, permissive hypercapnia, and pressure limited ventilation with positive inspiratory pressure limited to <40 cm H2O may also improve outcome.22 23 High-frequency oscillatory ventilation may be of benefit for patients with ARDS.24 Other therapies which have been used include steroids,25 prone positioning, inhaled nitric oxide, exogenous surfactant and extracorporeal membrane oxygenation. Mortality from pneumococcal pneumonia is between 2% and 4%.26
Pneumococcal sepsis
Blood stream infection is common in cases of IPD, either alone or in combination with focal disease. It is not within the scope of this article to describe the management of severe sepsis in detail. However, patients with signs of shock should receive standard ABC management according to American College of Critical Care Medicine–Pediatric Advanced Life Support (ACCM-PALS) guidelines27 with the focus on fluid resuscitation, early inotropic support and intubation and mechanical ventilation before clinical signs of pulmonary oedema develop. This is normally after the first 40–60 ml/kg of volume resuscitation (figure 6). There is good evidence that aggressive early fluid resuscitation and compliance with the ACCM-PALS guidelines improves outcome in paediatric septic shock28 and that failure to reverse shock by PICU admission is associated with increased mortality.29 Some children require large volumes of fluid over a short period of time to restore their circulating volume. There are no trial data to support the use of one fluid over another but it is common practice to use 4.5% human albumin solution after the first crystalloid bolus. Management should be determined in conjunction with the regional PICU. Early referral is recommended.

Current American College of Critical Care Medicine–Pediatric Advanced Life Support (ACCM-PALS) guidelines for management of septic shock.27 IV/IO/IM, intravenous/intraosseous/intramuscular; CVP, central venous pressure; FATD, femoral artery thermodilution technique; Hgb, haemoglobin; MAP, mean arterial pressure; PICCO, pulse contour cardiac output; PICU, paediatric intensive care unit ; ScvO2, central venous oxygen saturation.
Pneumococcal haemolytic uraemic syndrome
Haemolytic uraemic syndrome (HUS) may complicate IPD. Pneumococcal-associated cases account for 14% of all HUS diagnosed in the UK.30 Pneumococcal pneumonia with empyema is the most common precipitating illness, but pneumococcal HUS is also associated with meningitis and isolated pneumonia with bacteraemia.31 Children with pneumococcal HUS often have severe renal and haematological disease, requiring dialysis and support with blood products for thrombocytopenia and anaemia. Although, historically, HUS had a bad prognosis, recent advances in intensive care mean that the overall prognosis is now good in children developing pneumococcal HUS, with the majority of children returning to normal renal function after a period of renal support.
The pathophysiology of pneumococcal HUS remains poorly understood, but the enzyme neuraminidase produced by S pneumoniae is thought to play a pivotal role. Neuraminidase cleaves N-acetyl neuraminic acid from the surface of red blood cells and endothelial cells, uncovering the Thomsen-Friedenreich (T) antigen. The majority of people possess naturally occurring IgM antibodies to the T-antigen therefore exposure of the T-antigen results in antibody-mediated endothelial damage and haemolysis (figure 7). This causes a thrombotic microangiopathy of renal vasculature, with or without tubular necrosis.31 32

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
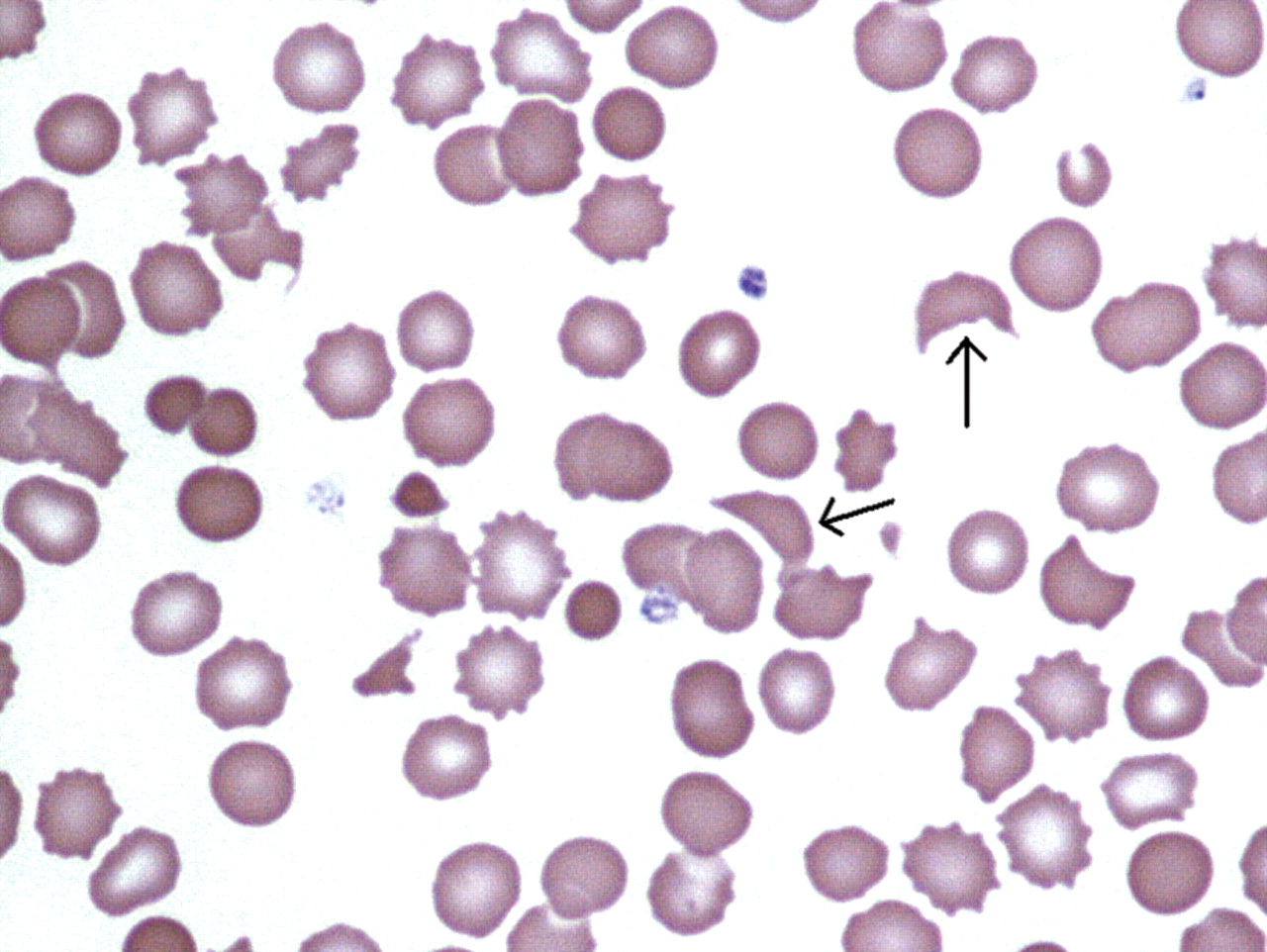{kind=link}
Peripheral blood film in haemolytic uraemic syndrome, showing schistocytes—erythrocyte fragments (marked) due to haemolysis, and thrombocytopenia.
Other IPD
S pneumoniae can cause suppurative infections in almost any part of the body. Abdominal infections such as peritonitis,33 ileitis and appendicitis have been reported, as have abscesses in solid organs. Peritonitis and ileitis are a particular risk in children with nephrotic syndrome. Other sites of reported infection include bone, joints,34 pericardium and placenta. These infections are rare complications and often occur in association with bacteraemia. Diagnosis can be difficult. Imaging, bacterial culture, antigen detection and PCR may facilitate diagnosis. Treatment principles include drainage of any purulent collections and antibiotic therapy, which should be guided by a specialist in paediatric infectious diseases. The duration of antibiotic therapy will depend on the response to treatment and the site of infection.
Further investigation of children who have had IPD
Most previously healthy children developing IPD do not have any underlying pathology. However, there are certain risk factors predisposing to IPD. Therefore, following a diagnosis of IPD, some thought should be given as to whether the child has an underlying condition increasing susceptibility to pneumococcus. Investigation may include testing for sickle cell disease and immune deficiencies, including HIV infection and hyposplenism.
Vaccination to prevent IPD
The first pneumococcal vaccines developed contained purified polysaccharide. However, such vaccines are ineffective in children under 18 months to 2 years, in whom purified polysaccharides are virtually non-immunogenic. To protect infants at highest risk of IPD, pneumococcal polysaccharide conjugate vaccines, eliciting T-cell help and effective from birth, have been developed.
The first such vaccine licensed in the UK, Prevenar (Prevnar, USA), contained seven serotype polysaccharides, (4, 6B, 9V, 14, 18C, 19F, 23F) conjugated to CRM197, a non-toxic recombinant variant of diphtheria toxin. After polysaccharide conjugate pneumococcal vaccine was introduced in the USA in 2000, cases of IPD fell by around two thirds in the high-risk under 2 age group,35 36 with vaccine serotypes falling by around 75% in the first 2 years.37 38 However, thereafter increasing numbers of cases of IPD caused by serotypes not included in the vaccination were reported, a phenomenon now known as serotype replacement.37 39 40 In 2010, 19A is now the most common serotype in the USA, representing 46% of the non-Prevenar isolates and currently 30% are multidrug resistant.41
Prevenar 7 was introduced in the UK in 2002 for ‘high-risk’ children <2 years.42 Coverage was extended to all children via the routine childhood immunisation programme at 2, 4 and 13 months from 2006. At this time there were around 5000 cases of IPD a year in the UK,43 more than 500 of these in children under 2 years old. In 2009 HPA data showed a good uptake of this vaccine, with 92.4% of children receiving the first two doses by 12 months, 86.6% receiving their booster by 24 months. On introduction of the vaccine, 76% of UK IPD isolates from children were serotypes included in the vaccine38 and the initial effect was gratifying, with substantial fall in reports of IPD.
The cumulative number of isolates of vaccine serotypes started to drop shortly after the introduction of the vaccine in 2006. This trend has continued to the present time, to the point of almost complete eradication. However, despite an overall fall in the number of cases of IPD following introduction of Prevenar 7, isolates of non-vaccine serotypes are increasing year on year, with a striking rise in serotypes 7F and 19A in the epidemiological year 2008–2009.38 To mitigate this problem, a 13-valent vaccine, Prevenar 13 (Pfizer, Tadworth, UK) has now been introduced in the UK, adding serotypes 1, 3, 5, 6A, 7F and 19A to the Prevenar 7 formulation. Kaye et al38 estimate this should increase present vaccine coverage by 50% against IPD. Unfortunately, ongoing serotype replacement makes for an unpredictable future. Surveillance will remain critical in the ongoing campaign against IPD as a cause of critical illness in children.
Acknowledgments
DI is grateful for support from the NIHR Biomedical Research Centre funding scheme. We thank Professor J Simon Kroll for his help in preparing this manuscript.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.