Article Text

Abstract
This paper describes a patient initially diagnosed with autoimmune thyroiditis who undergoes a diagnostic and investigatory journey
Statistics from Altmetric.com
Emily, a 15-year-old girl, with no previous medical history of note, presented to her general practitioner with symptoms of lethargy, dizziness and waking with nausea. Her general practitioner examined her and found her weight 60 kg (<75th centile), height 162 cm (25–50th centile) and body mass index (BMI) 22.9 kg/m2. Her cardiovascular and respiratory examinations were both normal, though she was noted to have mild epigastric pain on abdominal examination. There was no pallor or goitre evident. She was commenced on a trial of domperidone for the nausea and epigastric pain. Blood tests were taken for full blood count, electrolytes, liver function test, glucose and thyroid function test (TFT). These came back showing deranged TFTs. Thyroid stimulating hormone (TSH) 14.05 mIU/l (0.3–5.6) and free thyroxine (fT4) 6.5 pmol/l (7.5–21.1). Thyroid peroxidase antibodies were positive. The remainder of the results were within normal limits for her age.
She was commenced on 100 µg of levothyroxine, and her thyroid function soon became euthyroid on treatment.
Lethargy is a non-specific symptom, which often affects teenage children. It is most often non-pathological. However, if it is out of character and a cause for concern, several conditions should be considered. Adolescence is a stressful time, and the majority of teenagers will become more fatigued than in previous years partly due to increased life stresses and increasing demands placed on them from school, peers, family and sleep deprivation.1 A differential of pathological conditions should be borne in mind. Several pathologies can be insidious and present with very subtle signs, and so, it can be beneficial to carry out routine blood tests looking for anaemia, chronic renal failure, hypothyroidism and to consider infectious (eg, Epstein–Barr virus) or emotional causes for the change in behaviour. In addition to the baseline bloods tested, gastroenterological conditions such as inflammatory bowel disease and coeliac disease could be considered as Emily described symptoms of abdominal pain and nausea; these were not tested for on this occasion. In this instance, a diagnosis of autoimmune thyroiditis was made based on her deranged thyroid function and positive antithyroid antibody test.
Comment
Autoimmune thyroiditis (which, if associated with a goitre, is called Hashimoto disease) is a chronic inflammatory disorder of the thyroid gland with lymphocytic and cytokine-mediated thyroid destruction. Thyroglobulin autoantibodies/thyroid peroxidise autoantibodies are present in 95% of cases of autoimmune thyroiditis. It affects female patients more than male patients, and this ratio difference increases with age. Autoimmune thyroiditis is more common in conditions associated with abnormal karyotype such as Turner's, Klinefelter's and Down's syndrome and as part of other autoimmune diseases.2 3 It usually results in hypothyroidism requiring replacement therapy with levothyroxine, although patients can remain euthyroid and need regular monitoring of their thyroid function.
Following her initial diagnosis, Emily started to develop panic attacks which were centred around eating. She had never eaten breakfast and became self-conscious about eating in school in front of her peers, so she began skipping lunch. This resulted in her eating only one meal a day. She began to lose weight. Over the following 4 months, she found school increasingly difficult and stressful. Four months after her initial presentation, she stopped attending school altogether because of anxiety and panic attacks. Her parents became increasingly concerned by her weight loss and her worsening anxiety. She was referred to a child and adolescent psychiatrist for review.
Eleven months after her initial presentation, Emily now weighed 40 kg (0.4th centile) and her height 165 cm (50–75th centile), giving her a BMI of 14.6 kg/m2. She was biochemically euthyroid throughout – TSH 0.7 mIU/l (0.4–4.0), fT4 22.9 pmol/l (10.3–24.5). Emily was very needle phobic and would not eat at all for days before her blood test, so these were kept to a minimum.
A joint decision was reached with Emily's parents, paediatrician and psychiatric consultants that the most likely diagnosis was anorexia nervosa and that she would require admission to a specialist eating disorder centre.
Emily was admitted to a specialist eating disorder centre as an inpatient. She was diagnosed as having some body image distortion in keeping with a diagnosis of anorexia nervosa. However, it was also noted that she had some atypical features such as extreme anxiety with panic attacks on eating and absence of “fat phobia”; the food choices she made were not generally low in fat.
Comment
Anorexia nervosa is a psychiatric disorder characterised by a deliberate and extreme loss of weight, body image distortion and morbid fear of weight gain.4,–,7 Weight is obsessively controlled by means of deliberate starvation, excessive exercising, vomiting/purging and laxative, diuretic and diet pill abuse. Higher rates of childhood onset obsessive–compulsive disorder and social phobia are associated with anorexia nervosa.4 There is a female preponderance with only 10% of cases of anorexia seen in males. The incidence of anorexia nervosa is increasing, and it appears to be affecting children at younger ages, where there is a more equal sex ratio. The prevalence in the UK is thought to be approximately 1% in women aged 15–30 years old. Mortality from anorexia is 20% and results from suicide and complications of starvation (table 1).5
Management involves an individualised program usually in the outpatient but occasionally in the inpatient setting. The aim is to restore weight by approximately 0.5–1 kg/week. It generally combines nutritional rehabilitation, medical intervention, psychotherapeutic treatment, psychosocial rehabilitation and family therapies. Close monitoring during commencement of re-feeding is required as re-feeding syndrome can develop which can be fatal. This involves close monitoring of vital signs and blood tests to monitor electrolytes, phosphate, magnesium and glucose (table 2).4
Diagnostic criteria for anorexia nervosa ICD 1018
Emily's initial examination found her to be hypotensive, BP 80/60 mm Hg, tachycardic, HR 105 bpm with cool peripheries and an axillary temperature, 35.4°C.
Bloods:
Emily was commenced on a weight restoration programme which consisted of hydrating her with 2 litres per day of fluid orally. Gradual increase in daily calorie intake from 400 to 2000 kcal. All inpatients were kept on a salt-restrictive diet.
Emily continued on levothyroxine 100 µg and was commenced on multivitamin tablets and in light of her anaemia was also commenced on ferrous sulphate 200 mg three times daily.
Three weeks after admission, Emily began to deteriorate. Her panic attacks appeared to worsen and were associated with hyperventilation. Her BP dropped further to 67/50 mm Hg, HR 124 bpm. She had abdominal pain, headaches, nausea, feeling faint and vomited twice. She was brought by ambulance to accident and emergency.
On arrival at accident and emergency, she was found to have a BP of 94/60 mm Hg, HR 80 bpm, RR 16 bpm, Sats 100% air and was afebrile.
On examination, she was noted to be emaciated but alert and orientated with a Glasgow Coma Scale of 15/15.
She was well perfused with a capillary refill of <2 s and had normal cardiovascular, respiratory and abdominal examination. She denied any abdominal tenderness during her examination (fig 1).

(A) Photo of Emily a year before the admission to hospital and (B) during hospital admission.
Routine bloods were taken and shown:
She was given a provisional diagnosis of symptomatic hyponatraemia secondary to drinking large volumes of water while on a no-salt diet. She denied diuretic and laxative abuse. The possibility of adreno-cortical insufficiency was also considered.
Emily was known to suffer from anorexia nervosa, and so, it was not unexpected that she should be underweight, hypothermic, hypotensive with hyponatraemia as this would all be compatible with her diagnosis. The hyperkaelaemia could be put down to blood sampling difficulties in a peripherally shut down anorexic patient causing haemolysis and raised potassium levels. Many people with hypotension especially if they have postural hypotension can feel nauseated and dizzy on standing up.
Comment
Hyponatraemia is a common biochemical abnormality and can be divided into four main categories: (1) sodium loss, (2) dilutional effect (water excess), (3) inappropriate antidiuretic hormone (ADH) secretion and (4) pseudo-hyponatraemia due to displacement of sodium to the aqueous phase but still being measured as part of the total volume of plasma. This can occur in states of hyperglycaemia/hyperlipidaemia or hyperproteinaemias. The actual plasma osmolarity of sodium is actually within normal levels and, hence, does not require correcting but should be calculated using specific formulae to ensure this (table 3).
Emily was put onto a strict input/output fluid and food chart and was restricted to 500 ml of fluid overnight and was encouraged to have salty foods, which she admitted she had been craving.
Her morning bloods showed:
Her levothyroxine was decreased to 75 µg, and in light of a low random cortisol level, a synacthen test was performed (table 4).
Synacthen test
On closer inspection postsynacthen test, it was noticed that Emily had increased pigmentation of her knees, knuckles and hands (fig 2).

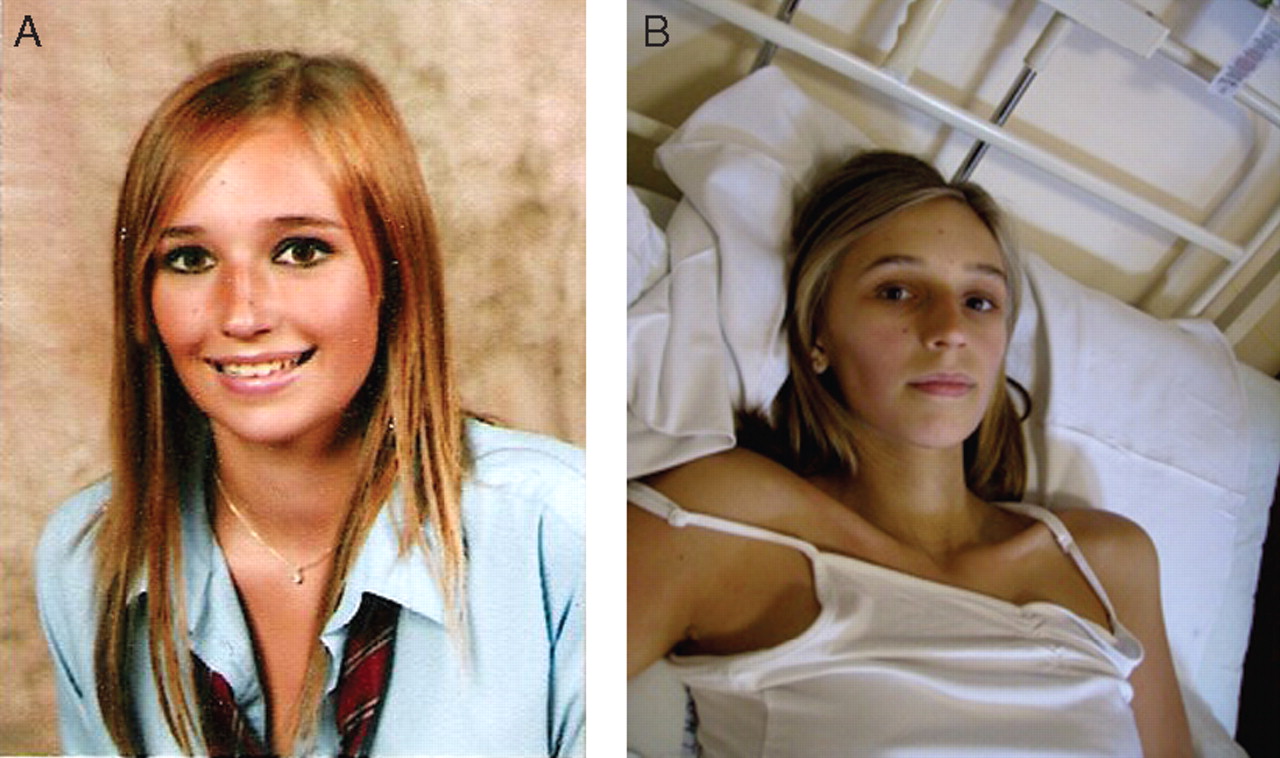{kind=link}
{kind=link}
(A–C) Hyperpigmentation of Emily's knuckles (A) and knees (B and C).
Emily was diagnosed with Addison's disease and was commenced on replacement doses of hydrocortisone: 10 mg morning, 5 mg afternoon and 5 mg evening.
Emily's BP remained low at 84/48 mm Hg, HR 85 bpm. She was retching, had vomited once and had epigastric abdominal pain. She was unable to eat or drink, as she felt very unwell and her fluid input had been minimal.
Emily was commenced on intravenous hydrocortisone, given three boluses of 0.9% sodium chloride 10 ml/kg and commenced on maintenance intravenous fluid 0.9% sodium chloride.
On review, Emily's BP improved to 109/67 mm Hg, HR 110 bpm. Repeat bloods showed a gradual increase of her sodium over the accompanying days. She was commenced on oral fludrocortisone following her adrenal crisis and was put back onto oral hydrocortisone once she returned to haemodynamic stability.
Over the next few weeks, the following results came back which confirmed the diagnosis of Addison's disease.
Comment
Addison's disease is a relatively rare disorder with an estimated prevalence of 120/million population; it is even rarer in the paediatric population.3 8,–,10 Addison's disease is primary adrenal failure. Autoimmune adrenitis is the most common cause of primary adrenal insufficiency post-infancy. In the UK, it accounts for 70–90% of cases. There may be positive adrenal antibodies present. Lymphocytic infiltrate cause destruction of the adrenal cortex, leading to failure of cortisol, aldosterone and adrenal androgen production. Cortisol levels are decreased with loss of circadian pattern associated with a persistent elevated ACTH level (Emily's ACTH levels were very elevated at 1918 ng/l). ACTH has an affinity for melanocortin-1 receptor; thus, excess of ACTH results in hyperpigmentation. Symptoms include lethargy, weight loss, gastrointestinal complaints (anorexia, nausea, vomiting and diarrhoea), increasing skin pigmentation especially in the skin creases, old scars and buccal mucosa and salt craving. Due to the slow progression of the disease, diagnosis is often delayed, and it may only be diagnosed during an Addisonian crisis when demand for cortisol cannot be met by the failing adrenal gland. Therefore, an appropriate stress response cannot be elicited leading to vomiting, drowsiness, hypotension, hyponatraemia, hyperkalaemia, hypoglycaemia and eventually collapse and coma. Diagnosis can be confirmed by an elevated level of ACTH and lack of increase of cortisol in response to synacthen test. Renin levels are high with a low/undetectable aldosterone levels as seen with Emily. Fasting glucose is low/normal.
Management of addisonian crisis11
Fluid replacement with 20 ml/kg 0.9% sodium chloride, initial intravenous bolus of 2 mg/kg hydrocortisone (max 100 mg) followed by a hydrocortisone infusion. Hypoglycaemia should also be corrected – if necessary intravenous dextrose infusion can be started to maintain normoglycaemia. Urea and electrolytes should be monitored 2 hourly, blood glucose and BP monitored hourly until normalised and stable. A strict input/output fluid balance should be kept as well as replacement of all fluid losses and correction of dehydration. Sodium should be corrected slowly and carefully to avoid osmotic demyelination syndrome (central pontine myelinolysis). Once no longer in crisis, the patient can be converted onto oral hydrocortisone (12–15 mg/m2 daily in three divided doses 2:1:1) and fludrocortisone (0.15–0.25 mg/m2 daily, single dose), sodium chloride supplements should be continued until stabilisation on fludrocortisone. Hydrocortisone levels should be doubled if patient becomes unwell to keep up with the body's requirement under stress. The patient should wear a medical alert bracelet in case of emergency and carry an adrenal insufficiency card in their wallet.
Autoimmune polyglandular syndrome, type II
Schmidt first described multi-endocrine organ autoimmune disease in 1926.12,–,14 His index patient had autoimmune thyroiditis and hypoadrenalism. The diagnosis is made when occurrence of autoimmune adrenal disease is found with concurrent autoimmune thyroid disease and/or type I diabetes mellitus (DM). Seventy-five per cent of autoimmune polyglandular syndrome, type II (APS2) involve thyroid and adrenal failure with no preference for which organ fails first. Approximately 10% of APS2 have a combination of all three (adrenal, thyroid and type I DM). Patients with APS2 are at increased risk of developing other autoimmune diseases (table 5). At onset, if APS2 is suspected, it is worth testing patients in case they are at risk of complete APS2 for adrenal cortex antibodies and 21-hydroxylase autoantibodies and consider performing a synacthen test if they have presented with type I DM. If they presented with Addison's disease, it is worth testing for islet-cell antibodies, autoantibodies to glutamic acid decarboxylase, second islet autoantigen and carrying out an oral glucose tolerance test which may reveal a subclinical impairment before the onset of type I DM. All patients with APS2 should be tested for thyroid autoantibodies if not yet checked. Regular screening for coeliac disease should be performed, but no regular screening is required for other related autoimmune diseases, although they should be borne in mind if symptoms of any start to develop.14
Only 1% of patients with autoimmune thyroid disease alone or type I DM develop adrenal insufficiency; therefore, screening for adrenal insufficiency is not indicated unless there is a strong family history or clinical suspicion. However, thyroid function and coeliac disease should be routinely checked for in patients with type I DM.
Prevalence of APS2 is thought to be between 1.5 and 4.5/100,000 population. It occurs most commonly in middle-aged women with an average age of onset of 35–40 years and female/male ratio of 4:1.
APS2 is thought to result from cell-mediated immune responses that attack the end endocrine organ. The genetic basis for APS2 is not yet known but appears to be linked to alleles within the HLA-DR3-carrying haplotype and a polymorphism of the cytotoxic T lymphocyte antigen-4 (CTLA4).15 APS2 may be the result of an autoimmune response to an external antigen in genetically susceptible people. Approximately 50% of patients with APS2 report a family history of polyglandular failure.
A note of caution and lessons to be learnt
Although hypothyroidism is a common occurrence, it can occur in combination with autoimmune failure of other endocrine glands such as seen in autoimmune polyglandular syndrome type II.13 16 17 If this is the case, by restoring a patient to a euthyroid state, it may precipitate adrenal failure and Addisonian crisis as it did in Emily's case. Hypothyroidism reduces cortisol clearance thus increasing the basal cortisol level; however, once hypothyroidism is corrected, cortisol clearance increases thus reducing circulating cortisol availability. Hypothyroidism reduces the body's metabolic rate, which reduces the body's cortisol requirements. By becoming euthyroid, the metabolic rate of the individual returns to normal, but the cortisol requirement cannot be matched by the failing adrenal gland; therefore, the patient goes into Addisonian crisis. Physicians should, therefore, be particularly suspicious of patients who deteriorate postcommencement of hypothyroid treatment or have one or more autoimmune endocrine diseases and develop non-specific/serious illness. A morning cortisol level with paired ACTH level should be checked to ensure it falls within normal range. Paired testing is done by taking both blood levels at the same time so they can be interpreted in relation to each other, such that if a morning cortisol is sufficiently high, then the ACTH should be suppressed in relation to this. If there is any doubt, then a synacthen test should be performed as the gold standard for diagnosing primary adrenal insufficiency.
Clinical messages
▶ Hypothyroidism is a common condition. Occasionally, it may be associated with other autoimmune conditions.
▶ If patients deteriorate rather than improve following initiation of thyroid replacement therapy, this should trigger further investigations for other conditions including primary adrenal insufficiency.
▶ In patients with both hypothyroidism and primary adrenal insufficiency, restoration of euthyroid state without correction of the adrenal insufficiency first can cause an increase in metabolic rate and push them into an addisonian crisis.
Footnotes
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Commissioned; externally peer reviewed.