Article Text

Statistics from Altmetric.com
Introduction
Cystic fibrosis (CF) is the most common inherited genetic disease in the Caucasian population, affecting more than 10 000 people in the UK. Discovered in 1989, the underlying pathology is a defect in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene on chromosome 7 and the subsequently produced protein. More than 2000 mutations have been identified.
Prior to the development of therapies targeting the CFTR protein, treatment has focused on preventing and treating the result of this defect including thick secretions, recurrent infections and failure to thrive. This is a significant treatment burden to the patient and family that continues lifelong.
This summary will provide an overview of small molecule precision medicines (CFTR modulators) and describe how their development is revolutionising CF care.
CFTR protein
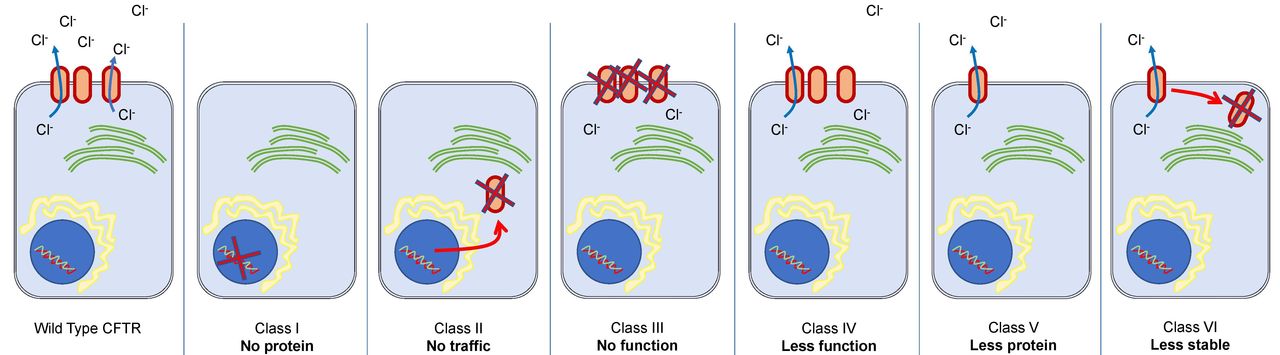
The CFTR protein is an ATP-dependent epithelial ion channel that contributes to the absorption and secretion of salt and water in various organs including the lungs, pancreas and gastrointestinal tract. Defects in the gene for the CFTR protein leads to malfunctioning or absent CFTR protein, which in turn leads to abnormal chloride conductance on the epithelial cell apical membrane. The precise mechanism of abnormal chloride transport varies depending on the specific gene mutation. The gene mutations have historically been grouped into six classes to represent the nature of the defect (figure 1).
{kind=link}
Representation of cystic fibrosis classification (I to VI). Class I—no protein: these mutations stop any recognisable CFTR protein from being produced due to stop codons in the gene. Class II—no traffic: these mutations affect CFTR processing in the endoplasmic reticulum (which recognises the malformed protein leading to protein destruction). Class III—no function: these proteins reach the cell surface, but the opening of the CFTR protein is affected. These are also called ‘gating defects’. Class IV—less function: this group of mutations reduce the passage of chloride ions through the CFTR protein channel, therefore reducing its function. Class V—less protein: these mutations result in a reduced amount of CFTR protein. Class VI—less stable: mutations here give a reduced amount of CFTR protein at the cell surface due to poor stability of the protein.
CFTR protein targeted molecules
The CFTR protein itself is an attractive therapeutic target as it is the underlying cause of the pathology in CF. Functional assays examining the activity of the chloride channel first needed to be developed to evaluate the efficacy of proposed new treatments. This then opened the door for pre-clinical work to examine the action of many different compounds in a short period of time, termed high-throughput screening.1
High-throughput screening is the concept of testing a large-scale chemical library of thousands of compounds in the search for therapeutic options. Using the simple and rapid assays that had been developed, investigators assessed the compounds, looking for ‘hits’—that is, identifying possible therapeutic options. Development can then continue with these compounds, moving towards those with clinical applications.
Ivacaftor (Kalydeco; Vertex Pharmaceuticals)
High-throughput screening of 228 000 drug-like compounds was performed using a fluorescence membrane potential assay designed to identify compounds that improved the likelihood of the CFTR protein achieving chloride transport. Substances that work in this way are known as CFTR potentiators. Ivacaftor is a CFTR potentiator and was the first small molecule drug to progress to use in clinical practice in CF. Its pharmacology was first demonstrated in cultured cells before its safety was established in humans. The first human trials examining drug safety and pharmacokinetics were conducted in adults from 2007 to 2008, and were initially short (14 to 28 days), but still, small improvements were noted in lung function (FEV1), with a big decrease in sweat chloride.2
The drug was then studied over longer time periods (up to 96 weeks) and, in order to use it safely in the paediatric population, was studied in younger patients; those aged 12 years and up from 2009, those aged 6 to 11 years from 2010, those aged 2 to 5 years from 2013 and those aged 1 to 2 years from 2016.3–6 At each stage, safety, tolerability and pharmacokinetics were examined to ensure parity with the adult studies. Studies are currently ongoing in even younger children.
Perhaps the most important step was the demonstration of the significant improvement in FEV1 (10.6%) in a large, randomised, double-blind, placebo-controlled trial in subjects with at least one G551D (a class III) mutation.3 (This large change surpassed previous gains in FEV1 from any other therapeutic agent in CF at that time.) Furthermore, a 55% relative reduction in the likelihood of pulmonary exacerbation was shown (important due to the association between exacerbation and rate of decline in lung function in patients with CF). This scientific breakthrough demonstrated that CFTR modulators could improve outcomes for children with CF.
In 2012, ivacaftor was licensed in the UK and is now used in people with at least one G551D mutation or one of several other gating (class III) mutations (therefore applicable only for a small percentage of the total CF population). EU approval has been recently granted for those aged 6 months or more.
Lumacaftor with ivacaftor (Orkambi; Vertex Pharmaceuticals)
The class II mutation F508del is the most common variation in the CFTR gene worldwide (with about 89% of patients with CF in the UK having at least one copy) and so is a clear priority for targeted therapy. The consequence of the F508del mutation is the misprocessing of the CFTR protein reducing its transport to the cell membrane.
Lumacaftor was discovered from a review of 164 000 small molecules and was demonstrated (during in vitro studies) to improve the trafficking of the F508del-CFTR protein to the cell membrane. Due to its mechanism of action, it is known as a CFTR corrector.
Clinical trials were performed with lumacaftor in combination with ivacaftor. Boyle et al demonstrated a modest reduction in sweat chloride and an improvement in FEV1 in patients over the age of 18 years who were homozygous for F508del. This provided evidence for the potential benefit of a combination CFTR modulator therapy.7
Larger studies of more than 1000 patients examined the change in FEV1 from baseline, demonstrating an improvement of 2.6% to 4.0%. The Cystic Fibrosis Questionnaire Revised (CFQ-R), an important patient-reported outcome measure, showed nominal significance. Studies in younger children followed showing encouraging signs of modification of the pathology including reduction in sweat chloride and an increase in faecal elastase.8
Since 2019, Orkambi has been nationally commissioned in the UK for use in patients with CF aged 2 years or older, who have two copies of the F508del mutation. Trials are currently underway in children aged 1 to 2 years.
Tezacaftor with ivacaftor (Symkevi; Vertex Pharmaceuticals)
Lumacaftor induces the CYP3A4 enzyme which can interfere with the metabolism of ivacaftor therefore limiting the use of lumacaftor–ivacaftor combination (Orkambi) in some patients. Another CFTR protein corrector, tezacaftor, was therefore examined in combination with ivacaftor (known as Symkevi in Europe). The improvement in lung function for those homozygous F508del was comparable with those seen in similar trials with lumacaftor–ivacaftor.9 10 Tezacaftor–ivacaftor was also examined in those patients heterozygous F508del with another mutation that provides some residual CFTR protein function (ie, a mutation that produces CFTR protein that retains some limited chloride transport activity) with similar results.11
In 2019, Symkevi was licensed in the UK for patients 12 years and over who have two copies of the F508del mutation, or a single copy with one of 14 ‘residual function’ mutations.
Tezacaftor with ivacaftor and elexacaftor (Kaftrio; Vertex Pharmaceuticals)
Kaftrio (Trikafta in the USA) is one of the next generation of modulators and is a triple combination therapy (made up of tezacaftor, ivacaftor and elexacaftor). It is highly anticipated, giving options to those patients who are heterozygous for F508del as well as to those who are homozygous. To increase the likelihood of developing a successful triple combination therapy, two investigational compounds (known as VX-445 and VX-659) were developed simultaneously, with VX-445 (elexacaftor) selected to move forward. Tezacaftor–ivacaftor–elexacaftor has recently been approved for use in patients over the age of 12 years, an achievement in record time.
A phase III trial for those over 12 years (heterozygous F508del with a second mutation with some minimal CFTR protein function) showed a very impressive increase in FEV1 of 13.8% (at 4 weeks) with a 63% reduction in pulmonary exacerbation compared with placebo.12 The CFQ-R score was 20.2 points higher in those treated versus placebo (a size of increase not seen in any previous study). A similar study in those over 12 years who were homozygous F508del showed impressive results with an increase in FEV1 of 10% when compared with tezacaftor–ivacaftor.13
Ongoing studies continue for children aged 6–11 years with a hope that similar efficacy is seen. Kaftrio is now licensed for use in those patients 12 years and older who are homozygous F508del or heterozygous F508del and a mutation that has ‘minimal function’.
Future outlook
The current selection of CFTR modulators potentially provides options for about 90% of patients according to genotype. Precision medicines are not currently available for the remaining 10% of people with CF in the UK. Ongoing research being undertaken by a number of different companies may provide similar targeted therapies for the rarer CF mutations.
A significant concern is access to these high cost medications. Complex and protracted negotiations preceded NHS access to some of these drugs. It is important to recognise the vital role cystic fibrosis charities and parent and patient groups have played in these negotiations. Prompt access to future therapeutic options must be a key priority for health leaders, in particular, when they have the potential to significantly change the outcomes for this condition in the long term.
The development of these small molecule medicines is revolutionising the care of CF and their use in younger patients gives hope for potential modification of the disease. There is already evidence that pancreatic function improves in children starting ivacaftor at a young age.
They are currently recommended as add-on therapies to the existing prescribed treatment and it is vital that these existing treatments continue to be adhered to by the patient. Apart from improving lung function, the significant reduction in pulmonary exacerbations will improve quality of life, reduce hospital admissions and potentially increase survival through reduced rate of decline in lung function. Real-world monitoring studies are underway to assess the impact of these new treatments.14 It will be necessary to carefully monitor these patients due to the unknown effect of this new class of medication, taken lifelong, after being commenced at a young age.
Overview of practicalities
Preparation and administration:
Granules available for ivacaftor and lumacaftor/ivacaftor up to age 6 years (could be mixed with cold food or liquid).
Tablets available for ivacaftor and lumacaftor/ivacaftor from 6 years and older, and tezacaftor/ivacaftor from 12 years and older.
Medication to be taken with a fat-containing meal/snack (with pancreatic supplementation if used) to aid absorption.
Standard dose: 12 hourly (dose adjustment required for interactions) (NB—tezacaftor/ivacaftor (Symkevi) is given in the morning with a further dose of ivacaftor only in the evening).
Baseline assessment and monitoring:
Liver function tests—performed at baseline, 3 monthly, then annually from 1 year from commencement of a new medicine.
Cataract screening—baseline assessment for cataract is required in children (<18 years) due to previous rare findings in earlier studies.
Side effects:
Medicines are generally very well tolerated. On initiation, transient upper respiratory symptoms and rash are the most common.
Significant side effects include abdominal pains and an increase in liver enzymes. Rarely results in discontinuation.
For further prescribing detail, please see the relevant sections for individual therapies online (www.medicines.org.uk/emc).
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @prasadnagakumar
Contributors CH, PN and MD contributed equally.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.