Article Text

Statistics from Altmetric.com
Introduction
PCRs have revolutionised the detection of bacteria in clinical samples since their widespread introduction in the 1990s.1 Quantitative PCR (qPCR), also known as specific PCR, involves the targeting of particular bacterial species. The technique uses specific primers (short strands of nucleic acid needed to initiate DNA replication) and fluorescent probes to allow real-time quantification of target bacterial DNA during amplification. The qPCR assay is a mainstay of microbiological diagnostics within the National Health Service (NHS). At our hospital approximately 200 qPCRs are performed per week for the investigation of bacterial infections. Although qPCR is by far the most frequently used molecular technique in bacterial diagnostics, in certain scenarios a broad-range (non-specific) 16S rDNA (ribosomal DNA) PCR is increasingly being used. Broad-range 16S rDNA PCR is also more commonly used in research settings, originally for use in detecting and identifying unusual bacterial species but now more widely used in the rapidly expanding field of microbiome research. This technique provides the initial step in the process of analysing complex microbial communities in human, zoological and even geological settings. In the future, analysis of individualised microbial communities using broad-range 16S rDNA PCR may be a key component of personalised medicine.
Quantitative or specific PCR
The two major benefits that qPCR offers over traditional culture methods are described below and in table 1.
Comparison of advantages and disadvantages of culture methods, qPCR and 16S rDNA PCR
Speed
Bacterial culture takes at least 24–48 hours for determina tion of a positive result, or longer for slow-growing organisms such as Mycobacterium tuberculosis.2 Furthermore, some fastidious organisms may commonly escape detection in routine cultures, for example, Kingella kingae.3 In qPCR, the use of fluorescent probes enables bacterial load to be detected and quantified in real time, hence reducing time to diagnosis and correct treatment initiation.4 The fluorescence is measured during the assay, and when it reaches a prespecified level the assay is considered positive. The number of cycles taken to reach this threshold is known as the cycle threshold value (CT value) and is proportional to the initial quantity of DNA present in the sample: that is, the lower the CT value, the higher the initial quantity of DNA (figure 1A). Strong positive results will be in the high teens or early 20s, whereas a sample with a CT value of 38–40 will be considered only borderline positive. Monitoring of the CT value can be used to assess therapeutic efficacy, for example, in tuberculosis.5
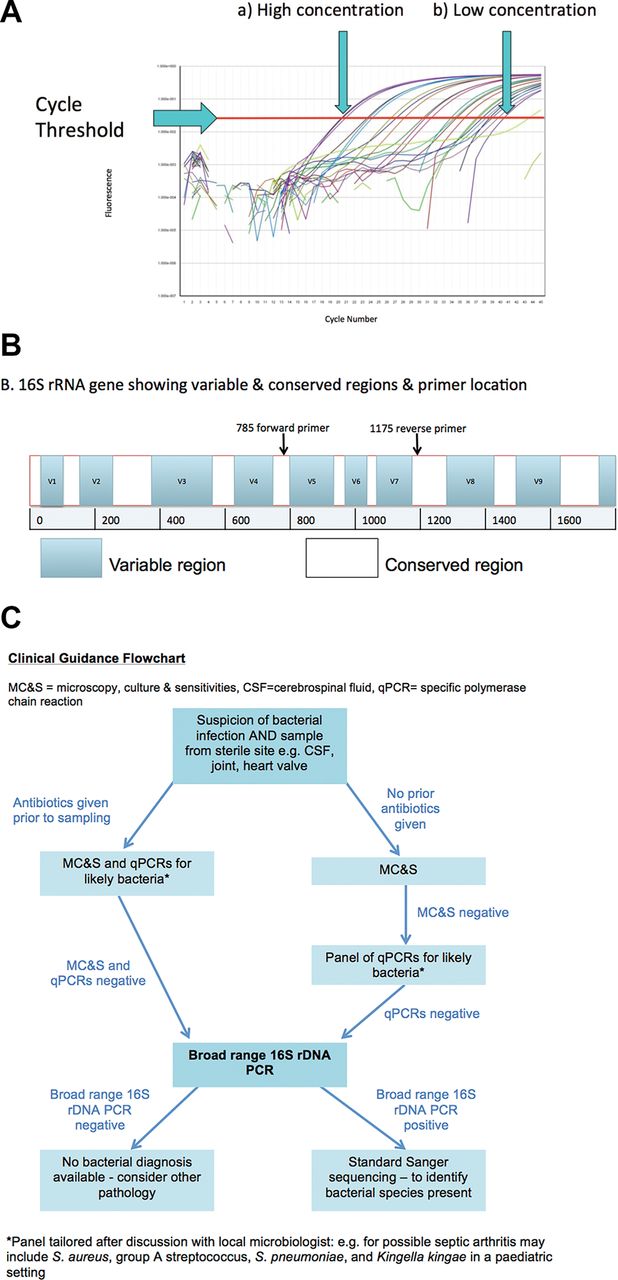
{kind=link}
(A) The result of a qPCR assay comparing fluorescence with cycle number. The results for multiple serial dilutions are presented demonstrating the cycle threshold value for each sample. The highest concentration sample (a) crosses the cycle threshold between cycles 20 and 21. The lowest concentration sample (b) crosses the cycle threshold at cycle 41. (B) Schema of 16S rRNA gene demonstrating variable (different between bacterial genera and species) and conserved regions (present in all bacterial rDNA) with placement of primers at conserved regions. After PCR amplification, the variable regions (V5–V7) are then sequenced to identify the bacteria present. (C) Flowchart demonstrating clinical guidance for the use of culture methods, qPCR and broad-range 16S rDNA PCR for sterile site infections where a bacterial cause is suspected. qPCR:quantitative PCR.
Utility despite prior antibiotic use
Culture only enables the detection of living organisms, and is therefore susceptible to false-negative results. qPCR will detect bacterial DNA regardless of viability, which is advantageous in a clinical context when empiric antibiotics may have already been given.6 7 This benefit also applies to broad-range 16S rDNA PCR (see below and table 1).
Broad-range 16S rDNA PCR
Unexpected or unusual pathogens may be missed if qPCR primers target only one bacterial species/family and qPCR testing panels may be necessary for diagnosis.8 Even with a large panel of qPCR assays, you will still only find what you set out to look for. Using a broad-range 16S rDNA PCR can address this issue. Primers are used that target the highly conserved regions of the 16S ribosomal subunit, present in all bacterial ribosomal genes (rDNA) (figure 1B). These subunits also consist of variable regions that differ between bacterial genera and species. Using broad-range 16S rDNA PCR, followed by DNA sequencing, allows the amplification and identification of any bacterial DNA present in a clinical sample.8 9 After sequencing, the sequences are compared with known nucleotide sequences on databases such as GenBank to identify the bacteria.8 10
Indications of broad-range 16S rDNA PCR
Broad-range 16S rDNA PCR can detect both viable and non-viable bacteria, similar to qPCR. It is also clinically useful when other techniques give negative results, for example, in culture-negative endocarditis, septic arthritis, meningitis or long-line infections.8 11 The bacteria identified are often unusual, rare, difficult to culture, or bacteria for which a specific PCR is not available.8 9 Examples include the identification of Helicobacter sp as the underlying cause of osteomyelitis or Neisseria meningitidis as the unexpected cause of septic arthritis by broad-range 16S rDNA PCR, after negative results were produced from other microbial diagnostic techniques.12 13
It is also possible to make distinctions between species: Ureaplasma spp consist of two bacterial strains, Ureaplasma parvum and Ureaplasma urealyticum, indistinguishable by culture, and each suspected to cause different pathology in neonates.7 14 15 Broad-range 16S rDNA PCR, followed by sequencing, has allowed the differentiation and identification of both species, aiding research into species-specific pathogenicity.8 16
Additionally, broad-range 16S rDNA PCR may identify previously uncharacterised bacteria. Broad-range 16S rDNA PCR enabled the identification of Bartonella henselae and Tropheryma whippelii as the pathogens underlying catscratch disease and Whipple’s disease, respectively.17 18
However, the breadth of broad-range 16S rDNA PCR renders it vulnerable to contamination. All bacterial DNA present in a sample is amplified, including that which is unavoidably present in reagents, meaning low-level environmental contamination is impossible to eliminate entirely. At a high number of thermal cycles, this low-level background contaminant DNA will be amplified and give a false-positive result. To reduce this risk, sequencing must be carried out to distinguish between a genuine pathogen and contaminants (often waterborne bacteria highly unlikely to cause disease). Using standard sequencing techniques, only the most dominant DNA sequence can be identified, which means that in samples where more than one bacterial species is present (such as stool) results are uninterpretable. This means broad-range 16S rDNA PCR with standard sequencing is not useful for samples from non-sterile sites.
To further reduce the risk of being overwhelmed by contamination, the number of thermal cycles is reduced compared with specific qPCRs, but this has the concomitant effect of reducing sensitivity.19 This means that broad-range 16S rDNA PCR will always be less sensitive than a well-designed specific qPCR to the order of 1–2 logs. A final disadvantage is that these methods may be limited to research or specialist laboratories, meaning samples may need to be sent away, increasing the turnaround time.16 A comparison of the key benefits and disadvantages between culture methods, qPCR and broad-range 16S rDNA PCR is shown in table 1, and a flowchart of suggested investigations for suspected bacterial sterile site infections incorporating both qPCR and broad-range 16S rDNA PCR is seen in figure 1C.
In summary, broad-range 16S rDNA PCR is a crucial adjunct to microbiological diagnostics as a second line when infection of a sterile site is highly suspected, but culture and qPCR for the most likely pathogens have been proven negative. Owing to the risk of detecting bacterial DNA contamination, fewer PCR cycles are performed than for qPCRs, resulting in a less sensitive assay, so qPCRs should be used first (figure 1C). In research, 16S rDNA PCR will continue to be used to identify novel bacterial species, characterise species-specific pathogenicity and as a gold-standard assay to compare against when evaluating new assays. It is also used in combination with cutting-edge techniques, such as next-generation sequencing. This is used to characterise complex microbial populations in the gut, stool, vagina, placenta, lung and in environmental settings.20 As next-generation sequencing based on broad-range 16S rDNA PCR becomes more affordable and widely available, these techniques have the potential to allow tailored therapy as our understanding of the complex interaction between ourselves and our microbial communities increases.21 For example, broad-range 16S rDNA PCR of microbial gut communities has identified distinct changes in conditions such as HIV, post preterm delivery of neonates and malnutrition.22–24 As we explore the potential therapeutic avenues revealed in these diverse conditions, it is likely that broad-range 16S rDNA PCR will be a cornerstone of further research.
References
Footnotes
Contributors FF and KAH came up with the concept. AP wrote the first draft. All authors contributed to the final draft.
Funding Dr Fitzgerald is supported by a grant from the Medical Research Council (MR/K023535/1) and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.