Article Text

Statistics from Altmetric.com
Introduction
Puberty is the physiological sequence of events when secondary sexual characteristics develop and reproductive capacity is achieved. The start of puberty is characterised by the appearance of a palpable breast bud (stage B2) in girls and testicular volumes of >3.5 ml in boys. These changes are brought about in response to the pulsatile secretion of gonadotropin releasing hormone (GnRH) from the hypothalamus stimulating the release of follicle stimulating hormone (FSH) and luteinising hormone (LH) from the anterior pituitary. It is still unclear why the GnRH neurons are usually inhibited after infancy until the start of puberty, although fat mass (via leptin signalling) and kisspeptin secreting neurons stimulating the GnRH system certainly play a role. LH acts on the gonads to increase the secretion of oestrogen in the female or nocturnal testosterone in the male that then promote growth in target tissues. FSH acts on the gonads to produce ovum or sperm, respectively. Androgens in particular promote other secondary sexual characteristics such as smelly feet and axillae, acne, body odour and mood swings – often most noticeable to the parents.
The timing of puberty is very variable and is dependent on socioeconomic status, ethnicity, nutritional and genetic factors. There has been an apparent secular trend in the onset of female puberty, although the mean age of menarche has remained fairly stable during the last 2 decades, implying that the duration of puberty may be increasing in girls.1 Hence, the definition of early or late puberty is fairly arbitrary. In the UK, the ranges for normal onset of puberty are shown on the growth charts,2 with the 98th centile for start of testicular enlargement just less than 9 years in a boy and just over 8 years for B2 in a girl. Similarly, only 2% of boys have not started testicular enlargement by 13.25 years of age and only 2% of girls have no changes regarding B2 by 13.75 years of age.
However, concern may be expressed by the patient and their parents about the timing of puberty at any age, especially in the context of genetic short or tall stature, and arrested puberty is always a sign of serious pathology. This article will consider the causes, investigation and treatment of early, delayed and arrested puberty.
Early puberty
In the UK, any pubertal changes before 8 years of age in a girl and 9 years of age in a boy are considered early, but this has to be interpreted in the light of family history, ethnicity, body mass index (BMI), social factors and past medical events. In some parts of inner city USA, breast development is not unusual at a much earlier age.3 Secondary sexual characteristics can develop in a concordant (testicular enlargement followed by penile growth with pubic hair; breast bud followed by pubic hair and later menarche) or discordant manner (enlargement of the penis with pubic hair but small testicles or breast development with no pubic hair, as two examples). Concordant puberty suggests central activation of gonadotropins (gonadotropin dependent precocity or central precocious puberty), whereas discordant changes (eg, progressive breast enlargement with no pubic hair, or penile enlargement with small testicles) suggest overactivity of sex hormone production in the periphery from the adrenal, gonads or tumours (gonadotropin independent). Peripheral testosterone (or other androgens) and oestrogen secretion will initially suppress LH and FSH secretion but eventually may activate the central pulsatility of gonadotropins, thus allowing central puberty to supervene.
The causes of central and peripheral early puberty are listed in table 1.
Causes of early puberty
Concordant, central precocity
By far the most common cause of early puberty is idiopathic early puberty in girls (at least 10 times more common than in boys). There are both aggressive and more indolent forms. There may be a history of early menarche in the mother in the slowly progressing form, which is commoner in the Afro-Caribbean or mixed race population, especially if the child is overweight and in children adopted to the UK from other countries.4 There will be breast and pubic hair growth coupled with a modest growth spurt and only slight (<1 year) bone age advance with few changes on pelvic ultrasound. In the more aggressive form, height velocity will be greater, bone age will be advanced more than 1 year from chronological age and the final height compromised in proportion to this advance (however height predictions based on bone age in early puberty should not be relied upon as they are wildly inaccurate).5 The mood swings in particular are troublesome for families. Both ovarian and uterine enlargement (>2 ml volume) will be evident on pelvic ultrasound and the uterus will be more angulated with respect to the vagina and become more pear-shaped. Oestrogen will be detectable along with elevated morning LH and FSH levels both at baseline and in response to stimulation.6 In boys with central gonadotropin activation, there will be testicular enlargement accompanying virilisation and again elevated LH/FSH and detectable testosterone, however idiopathic early puberty is much less likely. In addition to measuring LH/FSH and testosterone or oestrogen, it is also worth checking thyroid function for the rare cases of long-standing hypothyroidism where elevated thyroid stimulating hormone (TSH) can mimic FSH and induce early testicular and ovarian enlargement, often with little other evidence of virilisation.
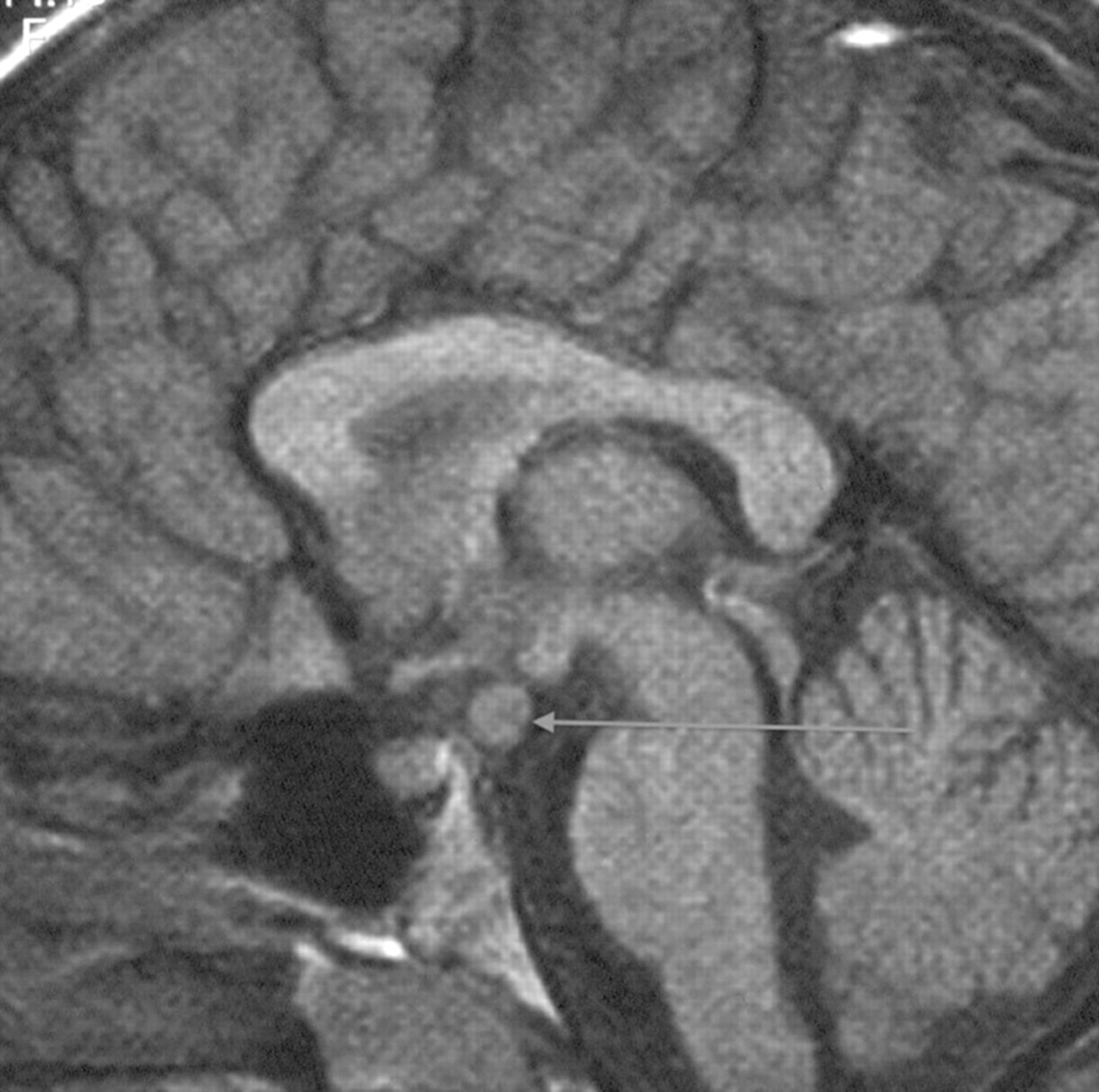
In the aggressive forms of early puberty, in girls less than 6 year of age and all boys, or if there are neurological symptoms, an MRI scan of the hypothalamus, pituitary and brain is required to exclude the second most common cause of central precocity in both sexes, that is, tumours around the pituitary stalk and hypothalamus (only about 10% girls compared to at least half of all boys presenting with central activation have detectable intracranial pathology). Most tumours except for optic gliomas in neurofibromatosis are hamartomas, but sequential scans are needed to prove lack of progression (figure 1). These hamartomas secrete cytokines that promote pulsatile gonadotropin secretion.7 Other genetic or acquired abnormalities of the brain can also produce central activation, as can low dose cranial irradiation in females when this is part of treatment for leukaemia.8
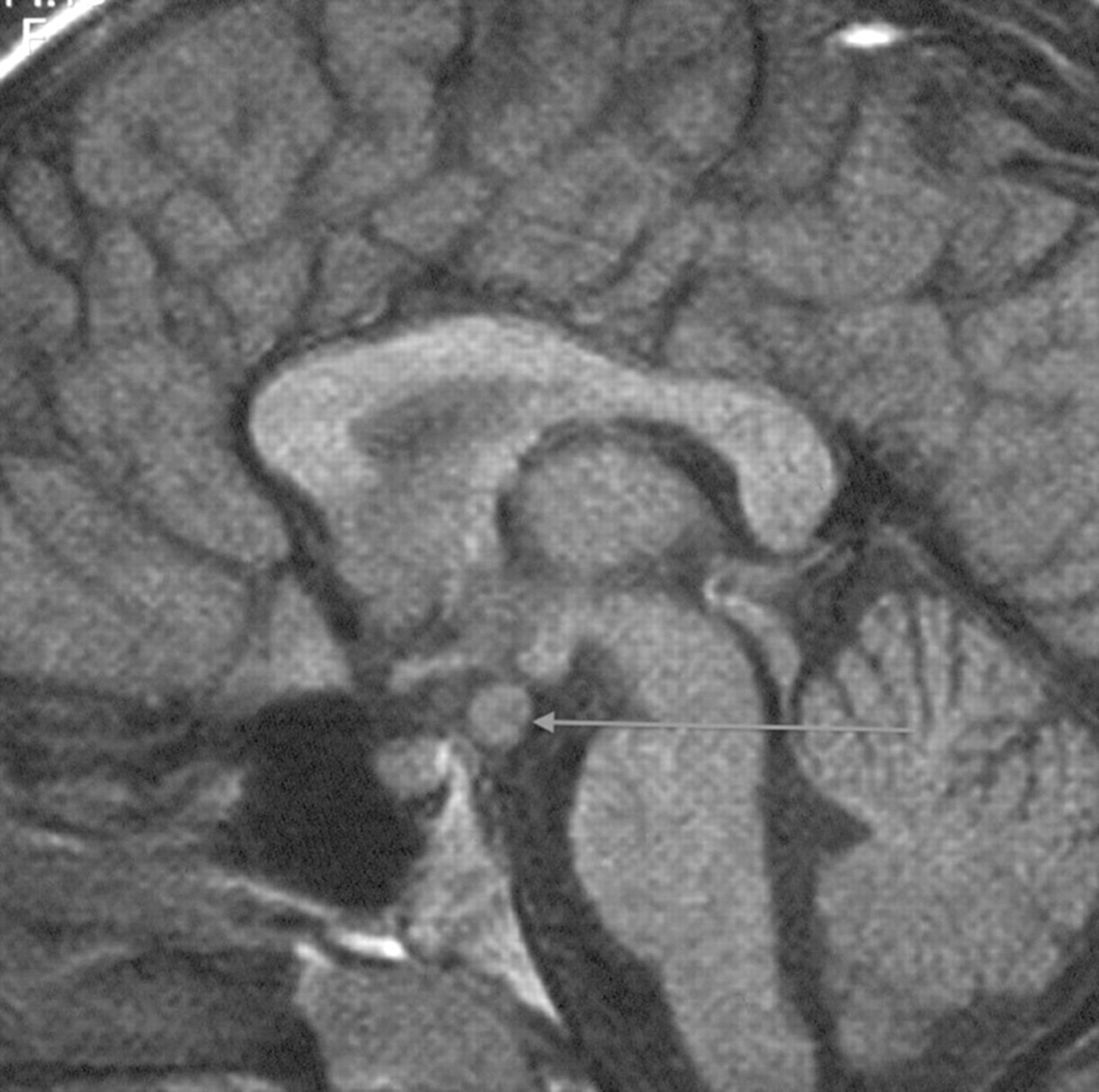
Saggital MRI showing hypothalamic hamartoma leading to central precocious puberty.
In all the above cases, the requirement for treatment needs to be discussed with the patient and their family. Good information booklets are available (http://www.bsped.org.uk). For genetically tall girls between 6 and 8 years of age with idiopathic early puberty unlikely to produce a reduced final height, it may be that no treatment is required except support to cope with periods and the behavioural and cosmetic changes. Approximately 10% of girls achieve menarche while at primary school, and there are better provisions for this than previously. However, the family may press for treatment to help reduce symptoms and delay menarche. If final height is likely to be reduced in the context of very early development or additional genetic short stature, then there are more ‘medical’ reasons for considering treatment. Treatment of central secretion of gonadotropins relies on the suppression of pulsatility by the use of long-acting GnRH analogues. A consensus statement regarding their use was published in 2009.9 They work by causing continuous release of LH and FSH from gonadotropes in the pituitary that can then no longer restock their supplies to produce further pulses of secretion. Therefore, it is sometimes worthwhile to cover the initial surge of LH/FSH release induced with the analogue by using a peripheral blocking agent, cyproterone, during initiation of treatment in children with precocious puberty presenting late and with severely reduced growth potential. Successful GnRH treatment relies on the continuity of sufficient dosage to prevent re-accumulation of LH and FSH; intermittent treatment, as used to be seen with intranasal GnRH agonists and sometimes is still seen if compliance is not assured, is harmful as it generates bursts of gonadotropin release. In general, it is easier to suppress puberty in boys than in girls, and many girls will escape from suppression before their next depot injection is due, requiring an increased frequency of treatment either to control symptoms or produce low LH/FSH levels just prior to their next injection. In girls with idiopathic central precocious puberty, the addition of Oxandrolone (a non-aromatisable anabolic steroid) or growth hormone to a GnRH analogue may result in improved final height.10
Human chorionic gonadotropin (HCG) is a molecule sharing the same α subunit as LH, FSH (and TSH) that can be produced from pineal, hepatic and testicular tumours. It acts mainly on the LH receptor, hence producing rather more androgens than would be expected for the testicular volume in boys. HCG and other tumour markers (eg, α fetoprotein) will be raised, but LH and FSH will be low and non-stimulable. A cranial MRI scan or CT/ultrasound to locate a peripheral tumour mass will aid detection, and treatment then relies on removal, chemotherapy or irradiation depending on the site of the lesion.
Discordant sexual development
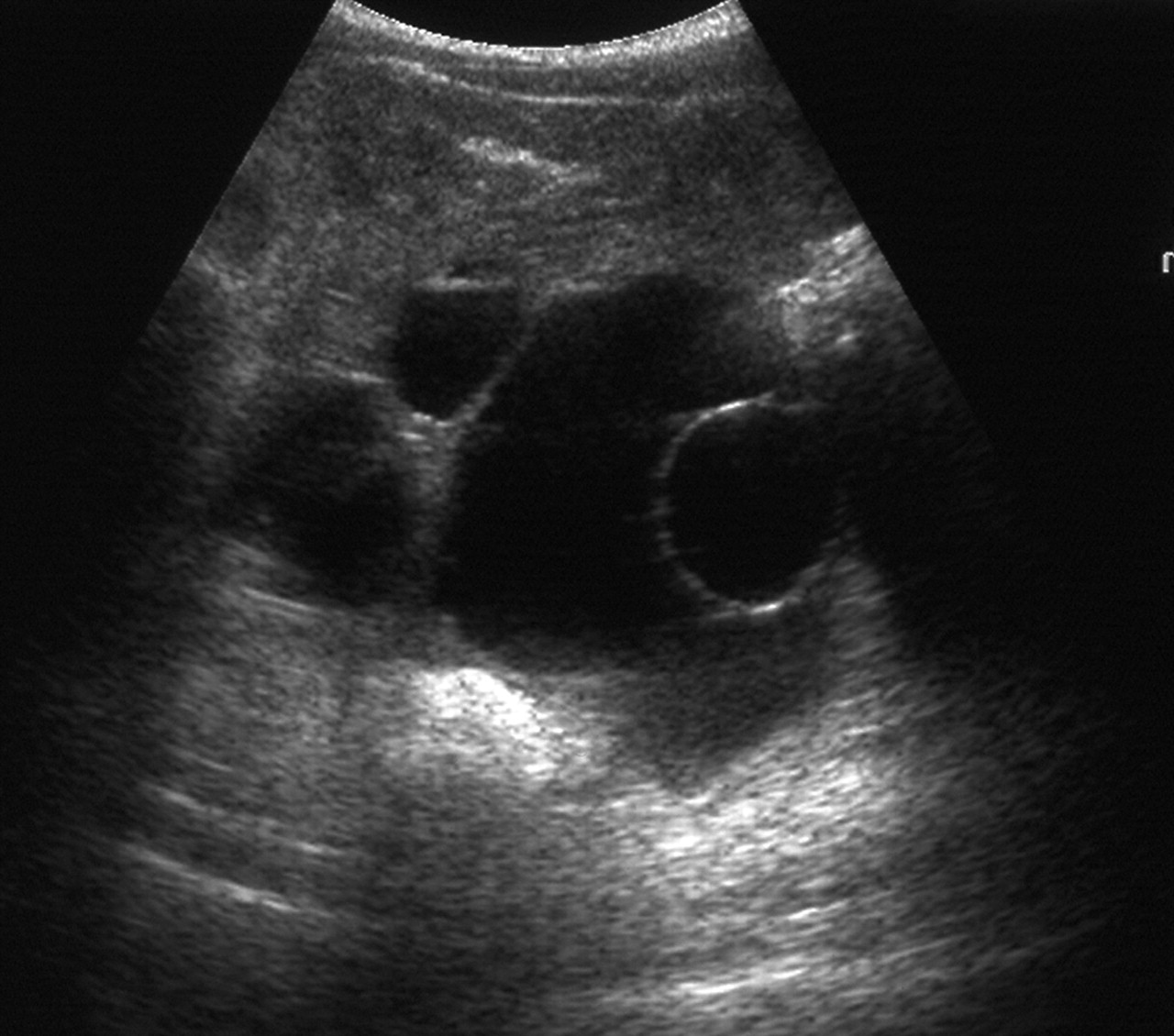
Thelarche is isolated breast development occurring in otherwise well children. It is commonly present from infancy, and although the breast size may wax and wane, it is non-progressive. It may be unilateral. Biochemical testing is usually normal and a pelvic ultrasound scan may show one or two large follicles in small volume ovaries11 12 (figure 2). No treatment is required. There is a slowly progressive variant, the ‘thelarche variant’, that is mid-way to central precocious puberty with a prominent FSH peak on stimulation testing.13 For the variant form, there is no good evidence that treatment is required or that this responds well to GnRH analogues.

Ovarian ultrasound through the bladder, demonstrating large cysts in a small volume ovary as seen in premature thelarche.
Ovarian (and adrenal, testicle or liver) tumours secreting oestrogen are rare and often present with a palpable mass and prominent breast development but no other signs of puberty. Gonadal tumours in either sex may also secrete testosterone, producing isolated androgenic effects. In massive ovarian oedema, subcritical torsion of the ovarian pedicle produces oestrogen (and sometimes testosterone) from the swollen ovary.14 With McCune–Albright syndrome, there are characteristic irregular lacy-edged café-au-lait patches, bony changes on x-ray and autonomous ovarian cysts (that may be huge; figure 3). In boys with the condition, there is penile and pubic hair growth, often more than would be expected from the testicular volume. In testotoxicosis, there is a similar genital finding but usually a male-limited family history of the disorder.
Ovarian ultrasound demonstrating autonomous oestrogen producing ovarian cysts in McCune–Albright syndrome.
Thelarche-like symptoms are produced from oestrogen containing medications and have been described after eating contaminated meat.15 Anabolic steroids produce androgenic effects and we have seen cases of virilisation of infants from transfer from the arms and hands of a father using testosterone gel, as also described by others.16
Adrenal androgens are the commonest cause of early virilisation leading to sexual hair growth (pubarche), body odour, acne, greasy hair and mood swings. If androgen levels are very high, there will be virilisation of the genitalia and a growth spurt with enhanced muscularity.
Adrenarche is a term used to describe normal maturation of the adrenal gland leading to enhanced secretion of the androgen dehydroepiandrosterone (DHEA). It is usually co-existent with the onset of normal puberty and contributes most of the androgenic component of puberty in young females. Precocious or premature adrenarche describes pronounced, early activation of DHEA production leading to androgenic changes before the onset of other pubertal changes. It may occur as a familial event, spontaneously or with an ill-understood association with hydrocephalus. There will be sexual hair growth, body odour, mood swings, some mild acne and greasy hair, and a small advance in bone age but no other virilisation. It requires no treatment, but families need to be advised that a proportion of girls affected will proceed to a polycystic ovary syndrome (PCOS) type phenotype, especially if they gain excessive weight through puberty.17
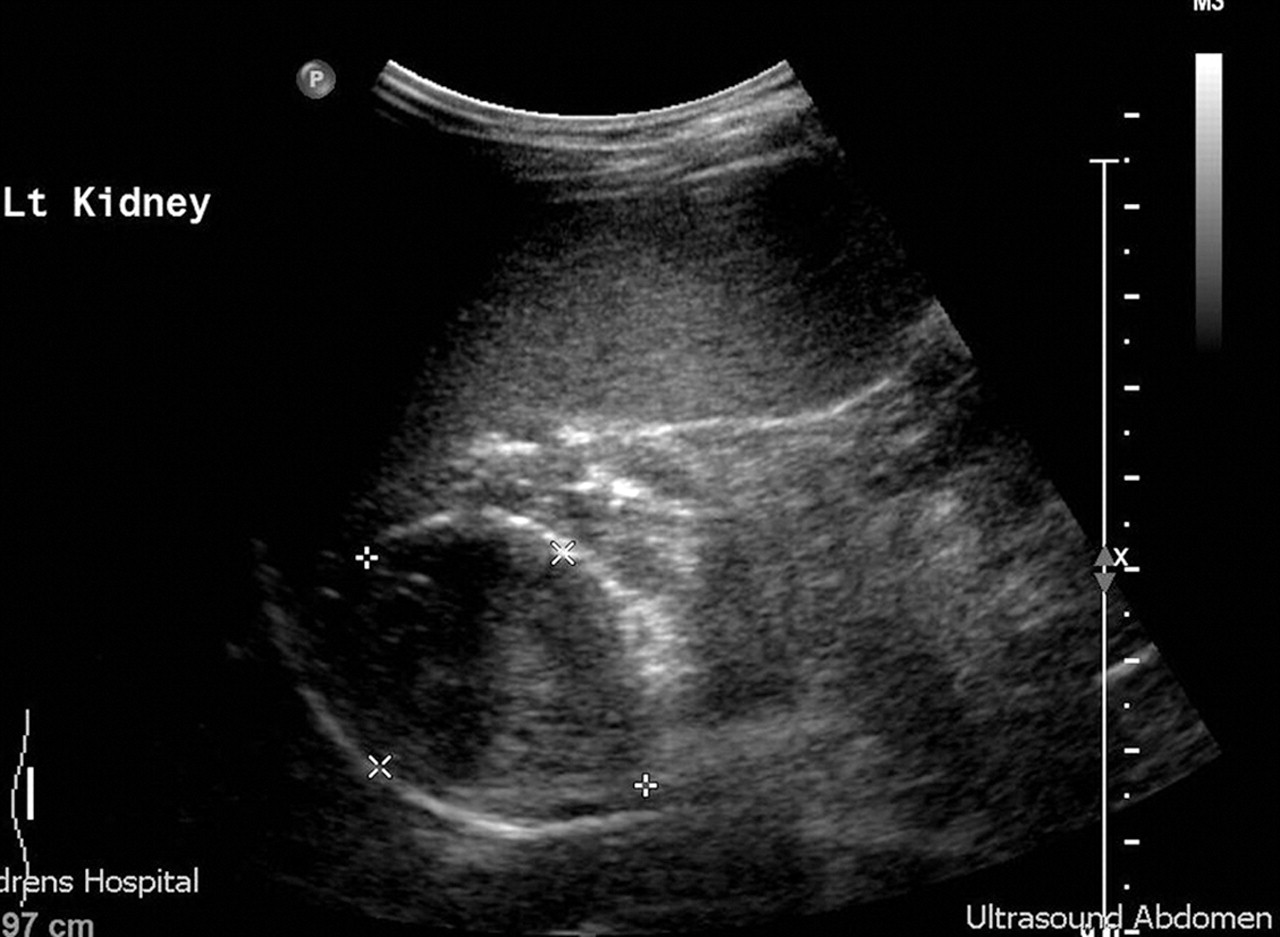
The mild form of non-salt losing atypical congenital adrenal hyperplasia (CAH) mimics adrenarche and can be easily differentiated by a urinary steroid profile or a short Synacthen test and measurement of 17 α hydroxyprogesterone. In more severe cases, virilisation is progressive and early, accompanied by a growth spurt and advanced bone age. Central gonadotropin secretion may then supervene with pubertal rises in LH and FSH on stimulation testing and requiring treatment with both hydrocortisone and GnRH agonists to achieve a reasonable final height. Adrenal adenomas and carcinomas (carcinomas tend to be larger at presentation) can secrete huge amounts of adrenal androgens. An abdominal ultrasound will detect the smaller adenomas (figure 4). Non-iatrogenic Cushing's syndrome in children tends to be accompanied by excess adrenal androgen secretion and hirsutism, occasionally with frank virilisation. Investigation of premature pubarche is outlined in box 1.
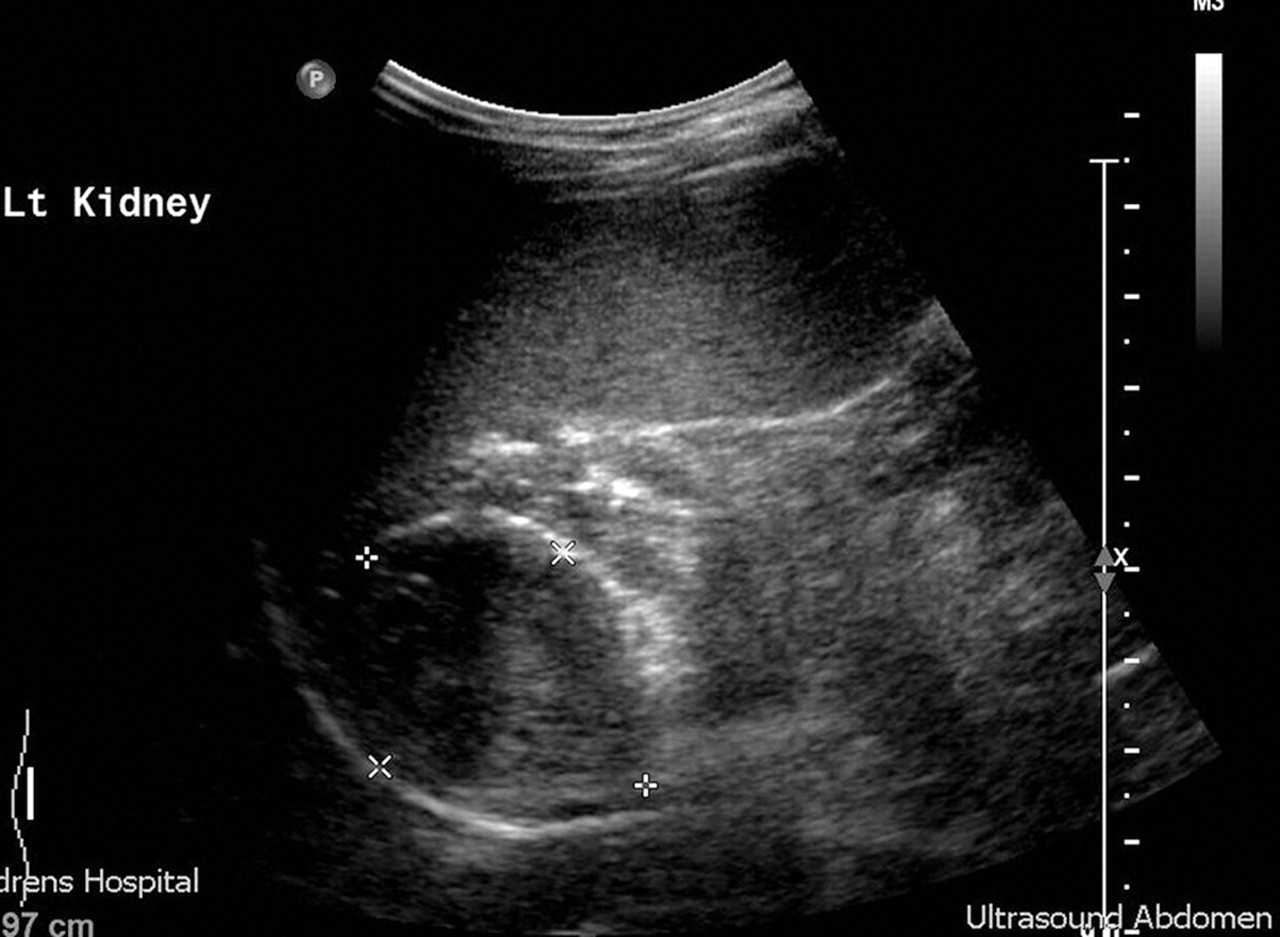
Abdominal ultrasound demonstrating androgen secreting adrenal adenoma. Carcinomas may be differentiated partially on larger size, but also on subsequent follow-up.
Box 1 Investigation of early pubarche
■ Testosterone, dehydroepiandrosterone (DHEA), androstenedione, 17 α hydroxyprogesterone OR urinary steroid profile
■ Abdominal and pelvic ultrasound
■ Bone age
Isolated premature menarche is a relatively common disorder of unknown aetiology. It typically presents in the summer months with two or three episodes of apparently cyclical bleeding in young girls with no other signs of puberty. Ultrasonography demonstrates a prepubertal uterus with no endometrial lining between bleeds. It is benign and non-progressive, but the differential diagnosis includes rare local lesions such as sarcoma, abuse and vaginal foreign body.
Some mild, transient breast enlargement occurs in approximately 50% of boys in early puberty, but severe persistent gynaecomastia presenting to paediatricians is increasingly common, possibly secondary to nutritional excess or environmental chemicals. It is not uncommonly unilateral and may then be related to chronic low grade trauma, for instance from a satchel strap. It usually accompanies early puberty but may occasionally be prepubertal. In many cases the appearance is worsened by obesity, possibly causing extra aromatisation of testosterone to oestrogen in the fatty tissue. Investigations for ectopic oestrogen secretion, karyotype, liver and thyroid function are usually normal. If present for more than 18 months, the tissue is relatively fixed and may require surgical removal with or without liposuction if worsened by subcutaneous fat in the pectoral area. If diagnosed early, then treatment with anti-oestrogen medication such as anastrozole may have some benefit.18
Delayed puberty
The history and examination in delayed puberty are often less helpful than the biochemistry. There may be a strong family history of late puberty, but this can also be the case in some pathologies causing delay. There will be no secondary sexual characteristics other than those that may arise from adrenal androgens (adrenarche). In boys the testicles will be 2–3 ml in volume, but smaller testicles are a clue to the diagnosis (eg, Klinefelter syndrome which may sometimes present with delayed puberty),19 as is associated micropenis (congenital hypopituitarism). Most children will be short and measurement of height and body proportions is very important as short stature for genetic height (Turner syndrome) and eunuchoid body proportions (simple delay and Klinefelter syndrome) may be present. An absent/blunt sense of smell is an important finding suggestive of Kallmann syndrome and schooling and behavioural problems may also indicate chromosomal abnormalities. Features to consider in the examination of cases of delayed puberty are given in box 2.
Box 2 Points to document in examination of delayed puberty
■ Height, weight, BMI, body proportions
■ Skinfold thickness if calipers available; in individuals monitored longitudinally, the relative loss of limb fat (sub-scapular skinfold now exceeding the triceps skinfold thickness) is another marker of the onset of puberty
■ Lanugo hair (sign of eating disorders), birth marks
■ Hernia repairs or other operative scars
■ External genital appearance: stretched penile length, imperforate hymen (primary amenorrhea); breast stage (and gynaecomastia in males)
■ Cryptorchidism or maldescent of testes; testicular volume
■ Dysmorphic features (Turner, Klinefelter, Prader–Willi syndromes, etc)
■ Signs of other disease (thyroid, CNS, inflammation)
■ Sense of smell (discrimination between three non-caustic smells)
BMI, body mass index; CNS, central nervous system.
However, late puberty can best be classified into two major groups based on the levels of LH and FSH. High, menopausal concentrations (hypergonadotropic hypogonadism) indicate primary gonadal failure and low concentrations (hypogonadotropic hypogonadism) indicate central disorders. Additionally, measurements of inhibin A and B (if available) and HCG may give further information about the function of the gonads in hypergonadotropic hypogonadism (box 3).20
Box 3 Investigation of hypergonadotropic hypogonadism
■ Karyotype (blood and, rarely, fibroblast/gonadal)
■ Pelvic ultrasound (uterine volume and ovarian size/morphology)
■ Abdominal MRI (intra-abdominal gonads)
■ HCG test with testosterone/oestrogen measurements (presence of secretory Leydig cells in ovary or testicle)
■ Inhibin B – presence of Sertoli cells or secretory follicles
■ Inhibin A – presence of corpus luteum
■ AMH – presence of testicular tissue (Sertoli cells)
■ Laparoscopy for biopsy, internal anatomy, orchidopexy or gonadectomy
■ Urine steroid profile (rare forms of CAH)
AMH, anti-Müllerian hormone; CAH, congenital adrenal hyperplasia; HCG, human chorionic gonadotropin.
Hypogonadotropic hypogonadism
The causes of hypogonadotropic hypogonadism are listed in box 4.
Box 4 Causes of hypogonadotropic hypogonadism
■ Constitutional delay
■ Structural abnormalities of the brain (tumours, infiltrations and infections)
■ Isolated LH and FSH deficiency (includes Kallmann syndrome)
■ Chronic disease
■ Hypothyroidism
■ Low BMI, eating disorders and excessive exercise
■ Rare single gene defects (DAX-1, LHR, FSHR, leptin and leptin receptor)
■ Prader–Willi, Laurence–Moon and Bardet–Biedl syndromes
BMI, body mass index; FSH, follicle stimulating hormone; FSHR, follicle stimulating hormone receptor; LH, luteinising hormone; LHR, luteinising hormone receptor.
By far the most common cause is constitutional delay, usually in healthy children who have delayed puberty, short stature with no growth spurt and a delayed bone age. Boys with simple delay present for treatment more frequently than girls and often have an affected father. They will show relative preservation of their leg length in comparison to their sitting height even before their puberty is noticeably delayed. Although they usually achieve a height in their genetic range, it is often at the lower end of the range.21
However, simple delay is often difficult to distinguish from isolated hypogonadotrophic hypogonadism, especially in males, and sometimes merges into this diagnosis with time. In both cases there are prepubertal levels of LH and FSH and even the results of stimulation tests can overlap. Only follow-up with or without treatment and sometimes re-testing will determine if puberty is going to occur spontaneously. Treatment is conveniently commenced by testosterone intramuscular depot injections of 62.5 mg (a quarter of a 250 mg vial) to 125 mg every 4 weeks. Alternatively, oral testosterone undecanoate 20–40 mg once a day may be used22 or, if available, Oxandrolone 1.25–2.5 mg once a day, which has the advantage that it may be started prepubertally to promote growth.23 If treatment is given for 3–6 months and then discontinued, follow-up will usually determine whether puberty spontaneously continues or stops. In permanent hypogonadism, treatment should be continued with escalating doses of testosterone over a 2-year period and a further period off treatment considered, with or without repeat testing, at 16 years of age when a 3-monthly depot preparation of testosterone can be used (Nebido 1000 mg). There is little experience with testosterone gels in paediatrics,24 but we have been using an escalating dose of Tostran gel (initially 10 mg, increasing to 60 mg a day in 10 mg increments) every 6 months to induce and maintain puberty in those not wanting injectable therapy.
In the fairly unusual case of a girl with simple delay requiring treatment, ethinyloestradiol 10 µg a day for 3 months may be used to initiate puberty. However, it is important to keep the course brief and to have excluded as far as possible any cases of permanent hypogonadism. Where prolonged treatment is needed, a low dose of oestrogen must be used initially (usually 2 µg), which is then increased gradually in 2 µg increments over the next 2–3 years to a final dose of 20–30 mg day with added progesterone for menstruation. Too rapid escalation of dose results in poor cosmetic breast development.25 Transdermal administration of oestrogen by the use of matrix oestrogen-only patches (which may be cut into quarters to administer doses as low as 6.25 µg transdermally for 3 days) is an alternative,26 with progesterone administration to induce menarche after 3 years, although some girls develop redness and irritation with the patches. Females with hypopituitarism often have sparse pubic hair from failure of adrenarche and this cosmetic problem can be treated with topical, low dose or injected testosterone or DHEA if required.27
Severe ill-health, especially with inflammation and steroid use, will delay puberty, as does a low BMI (<17) secondary to eating disorders and over-exercise.28 Treatment of the primary disease or nutritional problem is paramount, but sometimes replacement testosterone or oestrogen is required to preserve bone health if there are no signs of spontaneous puberty approaching 16 years of age.29
Rare single gene defects including DAX-1 (adrenal hypoplasia congenita) and mutations of LH/FSH and leptin (and their receptors) can also cause hypogonadotropic hypogonadism requiring life-long treatment.30 31
The fusion of the epiphyses is an oestrogen dependent process in both sexes.32 In males this is caused by aromatisation of testosterone. An improvement in predicted adult height of up to 5.5 cm in boys with constitutional delay in growth and puberty and idiopathic short stature using the aromatase inhibitor letrozole with testosterone in comparison with testosterone alone has been demonstrated.33 This experimental suppression of oestrogen biosynthesis could influence risk factors for cardiovascular disease, however, and long term risks and benefits may become more apparent with time.34
Hypergonadotropic hypogonadism
The causes of hypergonadotropic hypogonadism are listed in box 5.
Box 5 Causes of hypergonadotrophic hypogonadism
■ Klinefelter syndrome
■ XXX females
■ Turner syndrome
■ Gonadal dysgenesis
■ Testicular or ovarian failure
■ Androgen insensitivity syndrome
■ Single gene defects
Gonadotrophin secretion starts to increase before normal puberty, however in gonadal failure, lack of feedback suppression results in a rapid rise in gonadotrophin concentrations (FSH>LH) from 8 to 9 years of age.
Klinefelter syndrome (XXY, XXXY and other variants) is the most common cause of hypergonadotrophic hypogonadism in males and often presents with slow pubertal progression rather than delayed puberty. The boys are often tall with relatively long legs, there is cryptorchidism or small firm testis, rather poor musculature and, later, gynaecomastia. Testosterone levels begin to rise at normal ages and may attain adult levels, but then diminish in association with increasing oestrogen–testosterone ratios. There are few external features in girls with an XXX karyotype, but they tend to be tall, with amenorrhea and infertility. Those cases of Turner syndrome not diagnosed because of coarctation, dysmorphic features and short stature, may present as pubertal delay or arrest (more common in mosaic forms) with elevated menopausal FSH levels. Similarly, in gonadal dysgenesis (XO/XY males; XX, XO/XY females) there may be failure of non-adrenarchal pubertal development. In all the above, it may rarely be necessary to take both blood and fibroblast specimens to exclude tissue mosaicism.
Androgen insensitivity syndrome in the 46XY individual may be due to androgen receptor gene mutations and is variable in severity, ranging from the complete female phenotype with sparse pubic hair and amenorrhoea to the under-virilised male presenting with hypospadias. Testicular or ovarian failure may be due to surgery, radiation, chemotherapy, cryptorchidism, trauma, testicular regression syndrome or galactosaemia, and autoimmune with anti-testicular/ovarian antibodies. Rare single gene defects (5α reductase deficiency, aromatase deficiency, FSH receptor and LH receptor) may all present with pubertal delay.30
Lifelong treatment of hypergonadotropic hypogonadism is usually required, although sometimes apparent complete ovarian failure following treatment for malignancy (or even Turner syndrome) can reverse with resulting unexpected pregnancy. The treatment schedule is very much the same as that for hypogonadotropic hypogonadism, as above, with gentle initiation of puberty followed by escalation of oestrogen or testosterone to adult levels over a 2.5–3-year period, as large doses of oestrogen given too quickly result in a poor cosmetic appearance of the breast.25 The paediatrician should be aware of oestrogen prescriptions not originating from a hospital pharmacy as dosage errors (micrograms to milligrams) have been reported35 (also personal communication). In Turner syndrome or other cases with inherent short stature, there has been a tendency to postpone sex hormone replacement in an attempt to maximise growth, but the recent national Turner syndrome trial in the UK shows that a combination of growth hormone plus Oxandrolone allows maximal growth and the ability to induce puberty at a more socially acceptable time.36
Arrested puberty
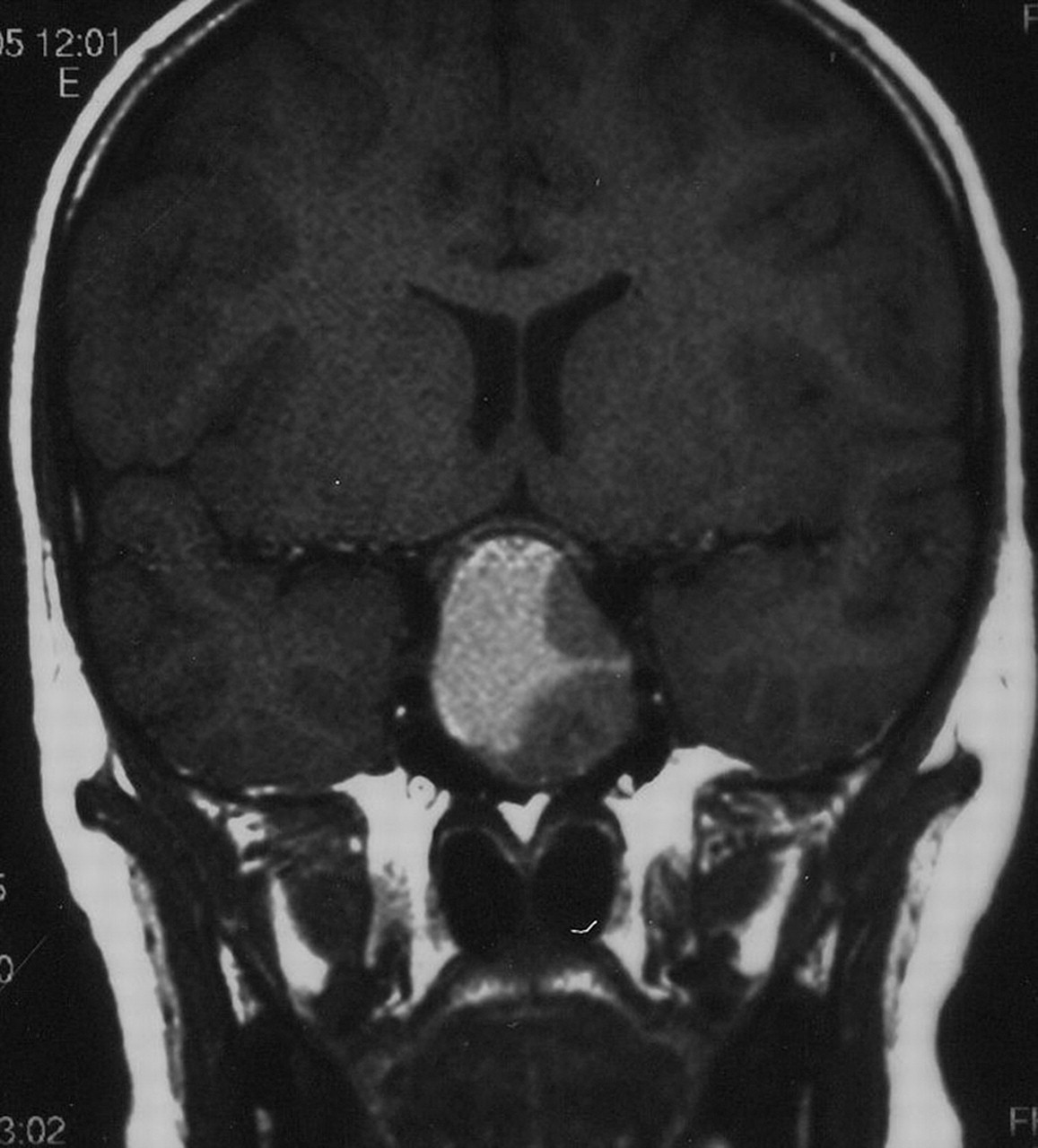
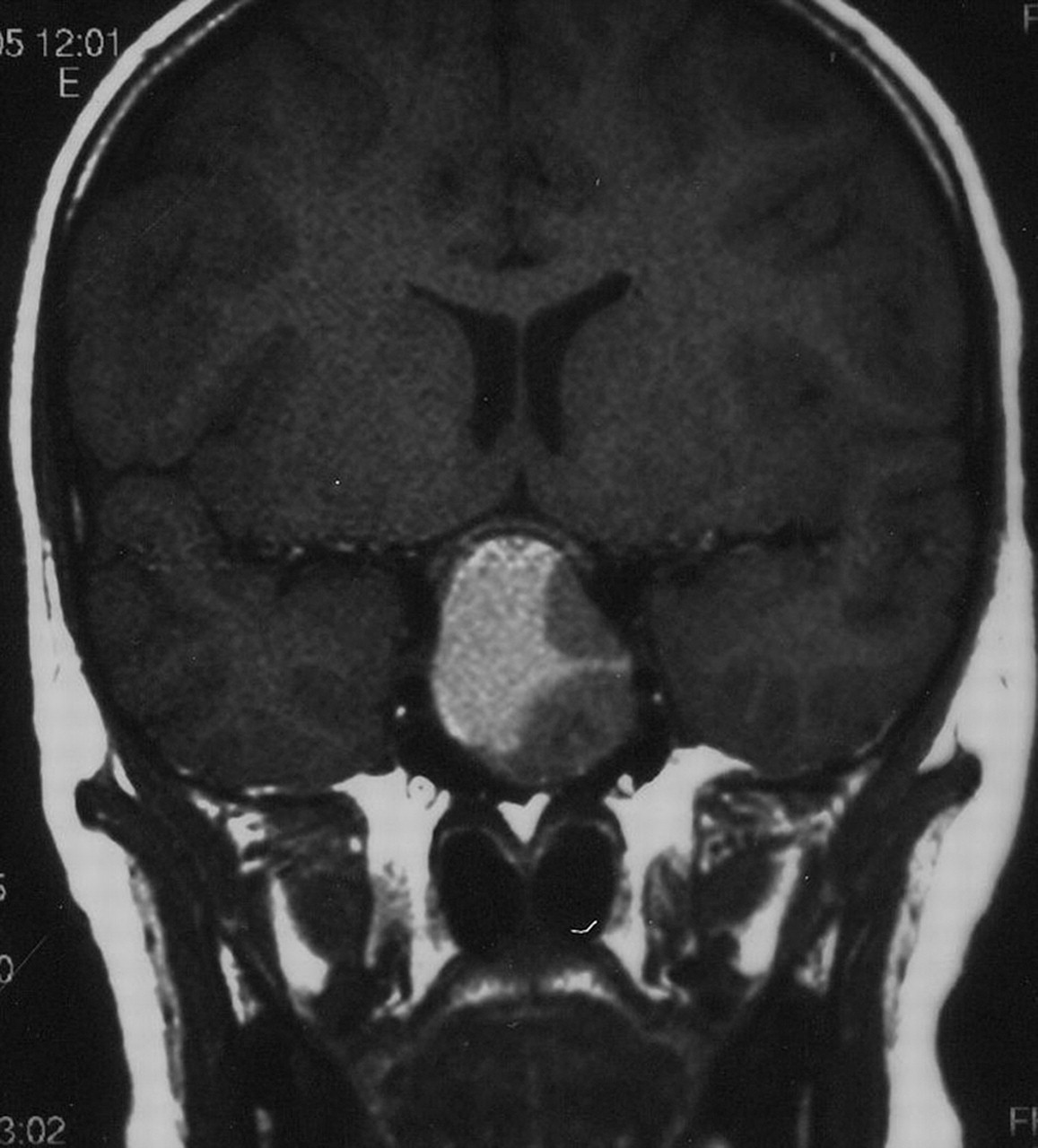
If puberty has commenced, but fails to progress, or some aspects regress, such as shrunken breasts or softening testicles, this implies an acquired failure of the pituitary–gonadal axis. In the case of severe ill-health or steroid administration the cause is obvious but otherwise requires urgent investigation.37 If the LH/FSH levels are low for the stage of puberty previously achieved, then hypothalamic or pituitary causes are most likely. The rest of the pituitary axis should be tested and MRI imaging obtained to exclude craniopharyngioma or other intracranial tumour. Pituitary destruction or damage can also rarely be secondary to head trauma, infection or autoimmune processes. Prolactinomas (figure 5) may present primarily with pubertal arrest, sometime associated with galactorrhoea, but always with an elevated prolactin level. Autoimmune hypothyroidism should be excluded.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prolactinoma presenting with arrested puberty; the prolactin level was 12 000 pmol/l (normal <800).
If the LH/FSH levels are menopausal, then a karyotype (with or without fibroblast samples) is required to exclude Turner mosaicism, androgen insensitivity and Klinefelter syndrome in particular. Gonads may be damaged by infections and autoimmune processes.
Premature menopause may occasionally present in the early teens with amenorrhoea after previously normal puberty. FSH is elevated and there is an increased likelihood of the patient carrying the gene for fragile X syndrome.38
Amenorrhoea or irregular periods are often due to polycystic ovary syndrome, which is an increasingly common condition linked with obesity and metabolic syndrome. Some girls with premature adrenarche evolve into the PCOS phenotype. PCOS is also more common in those who had intrauterine growth retardation as infants.39 In addition to usually a increased BMI, there is often acanthosis nigricans indicating insulin resistance. LH levels are typically higher than FSH levels. Because of the high insulin levels, the sex hormone binding globulin levels are very low, leading to a raised free testosterone and hirsutism with amenorrhoea or oligomenorrhoea. Treatment of PCOS is primarily through weight loss and lifestyle changes. Anti-androgens, such as cyproterone acetate, may be indicated in severe cases and metformin can possibly help restore regular menstruation.40 Cosmetic measures such as depilation or the use of eflornithine to reduce facial hair may help self-image.
Summary
Pubertal disorders are common and cases of both early and late puberty will present to all general paediatricians.Simple delay of puberty, certainly in the male, can be managed outside the specialist setting as long as the investigations outlined above are performed and treatment is given for a brief period followed by reassessment.Sexual precocity can safely be investigated to identify the simple disorders of thelarche and adrenarche which require no treatment. Other causes of sexual precocity and delay need investigation and treatment at a tertiary endocrine unit.41
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.