Article Text

Abstract
Controlled fasts can play a valuable role in the diagnosis and management of hypoglycaemia in paediatric clinical practice, but are no substitute for the collecting of appropriate critical samples at the time of hypoglycaemia for metabolic and endocrine studies. Fatty acid oxidation defects, hyperinsulinism and adrenal insufficiency should always be excluded prior to organising controlled fasts. Controlled fasts are safe if conducted in an experienced setting with strict protocols in place. Failure to adhere to protocol can defeat the purpose of the study and can potentially be dangerous. Proper planning in conjunction with the laboratory and close supervision by staff experienced in controlled fasts is crucial to ensure the best quality information is yielded from these procedures.
- hypoglycaemia
- controlled fast
- fasting tolerance
- Idiopathic ketotic hypoglycaemia
Statistics from Altmetric.com
Introduction
Hypoglycaemia is not infrequent in paediatric clinical practice and can potentially result in significant morbidity and mortality.1–3 Some metabolic and endocrine disorders can cause recurrent hypoglycaemia in childhood and these might be potentially preventable.
Controlled fasts are used to evaluate hypoglycaemia safely in a hospital setting. The aim of this article is to help paediatricians understand the role of controlled fasts in the investigation and management of hypoglycaemia.
What is the definition of hypoglycaemia?
There is no agreement between paediatricians and among paediatric textbooks about the lowest safe blood glucose levels in childhood. The definition of hypoglycaemia should ideally be based upon the lowest levels at which normal neurological function is affected. This varies between individuals, and depends on several factors including the rate of fall in blood glucose, the properties of the glucose transporter GLUT1 (which transports glucose across the blood-brain barrier), variation in individual neural susceptibility to low glucose concentrations and the ability of neurons to use ketone bodies for energy production. Nevertheless, it has been shown that neurological dysfunction in newborns and children does not occur until plasma glucose levels fall below 2.6 mmol/L,4 ,5 and this can be used as reasonable value for defining significant hypoglycaemia in childhood. In practice, we recommend prompt treatment of hypoglycaemia if plasma glucose falls below 3 mmol/L in asymptomatic patients and in patients with symptomatic hypoglycaemia regardless of the result.6–9
Metabolic and endocrine disorders causing hypoglycaemia in childhood (boxes 1 and 2) and tests recommended for investigation of hypoglycaemia (table 1) are outlined below.
Metabolic causes of hypoglycaemia in childhood
Glycogenoses
Glycogen storage diseases (I, III, VI, IX)
Glycogen synthase deficiency (GSD 0)
Glucose transporter defects
Fanconi-Bickel syndrome
Gluconeogenesis disorders
Fructose-1,6-bisphosphatase deficiency
Glucose-6-phosphatase deficiency (GSD 1)
Fatty acid oxidation disorders
Primary carnitine deficiency and carnitine cycle defects
MCADD, LCHADD, VLCADD
Multiple acyl-CoA dehydrogenase deficiency
Ketone synthesis defects
HMG-CoA synthase and lyase deficiencies
Ketone utilisation defects
SCOT, ACAT1, MCT1 deficiencies
Hereditary fructose intolerance
Mitochondrial respiratory chain defects
Liver disease (eg, galactosaemia)
Other conditions associated with hypoglycaemia: tyrosinemia type 1 and organic acidaemias (not common)
Ghosh et al.9
ACAT1, acetoacetyl-CoA thiolase; HMG-CoA lyase, hydroxymethylglutaryl-CoA lyase; HMG-CoA synthase, hydroxymethylglutaryl-CoA synthase; LCHADD, long chain hydroxy acyl-CoA dehydrogenase deficiency; MCADD, medium chain acyl-CoA dehydrogenase deficiency; MCT1, monocarboxylase transporter 1; SCOT, succinyl-CoA oxoacid transferase; VLCADD, very long chain acyl-CoA dehydrogenase deficiency.
Endocrine causes of hypoglycaemia in childhood
Hyperinsulinaemic hypoglycaemia
Hyperinsulinism-hyperammonaemia syndrome
Congenital hyperinsulinism
Dumping syndrome
Insulinoma
Beckwith-Wiedemann syndrome
Exogenous insulin (diabetes, factitious hyperinsulinism)
Adrenal insufficiency
Primary adrenal insufficiency
X-linked adrenoleukodystrophy
Withdrawal of exogenous corticosteroids
Hypopituitarism
Growth hormone deficiency
Hypopituitarism
Isolated growth hormone deficiency
Other causes
Idiopathic ketotic hypoglycaemia
Ghosh et al.9
Tests recommended for investigation of hypoglycaemia/hypoglycaemia screen
Physiological background
Normal adaptive mechanisms during fasting
During fasting, blood glucose levels are sustained as a result of adaptive hormonal and metabolic counter regulatory mechanisms.
In the early fasting state, insulin levels decrease and levels of the counterregulatory hormones glucagon, growth hormone and cortisol increase. Glucagon stimulates glycogenolysis to release glucose from liver glycogen. When glycogen stores are depleted, glucose is formed from amino acids and glycerol by gluconeogenesis.
Insulin inhibits lipolysis which is activated in the fasting state to release free fatty acids, which are subsequently β-oxidised resulting in the generation of ketones. An increase in metabolites such as free fatty acids may precede hypoglycaemia. Ketone bodies, namely acetoacetate and 3-hydroxybutyrate are used as an alternative source of energy.10
The occurrence and intensity of ketonaemia is proportionate to the extent of hypoglycaemia.10 ,11
In the fed state, insulin levels increase which switches off lipolysis leading to a fall in the level of ketone bodies.
A typical example of the metabolic changes seen during a prolonged fast in a 3-year-old child is shown in figure 1.

Typical metabolic responses to a 20 hour fast in a normal 3 year old boy.
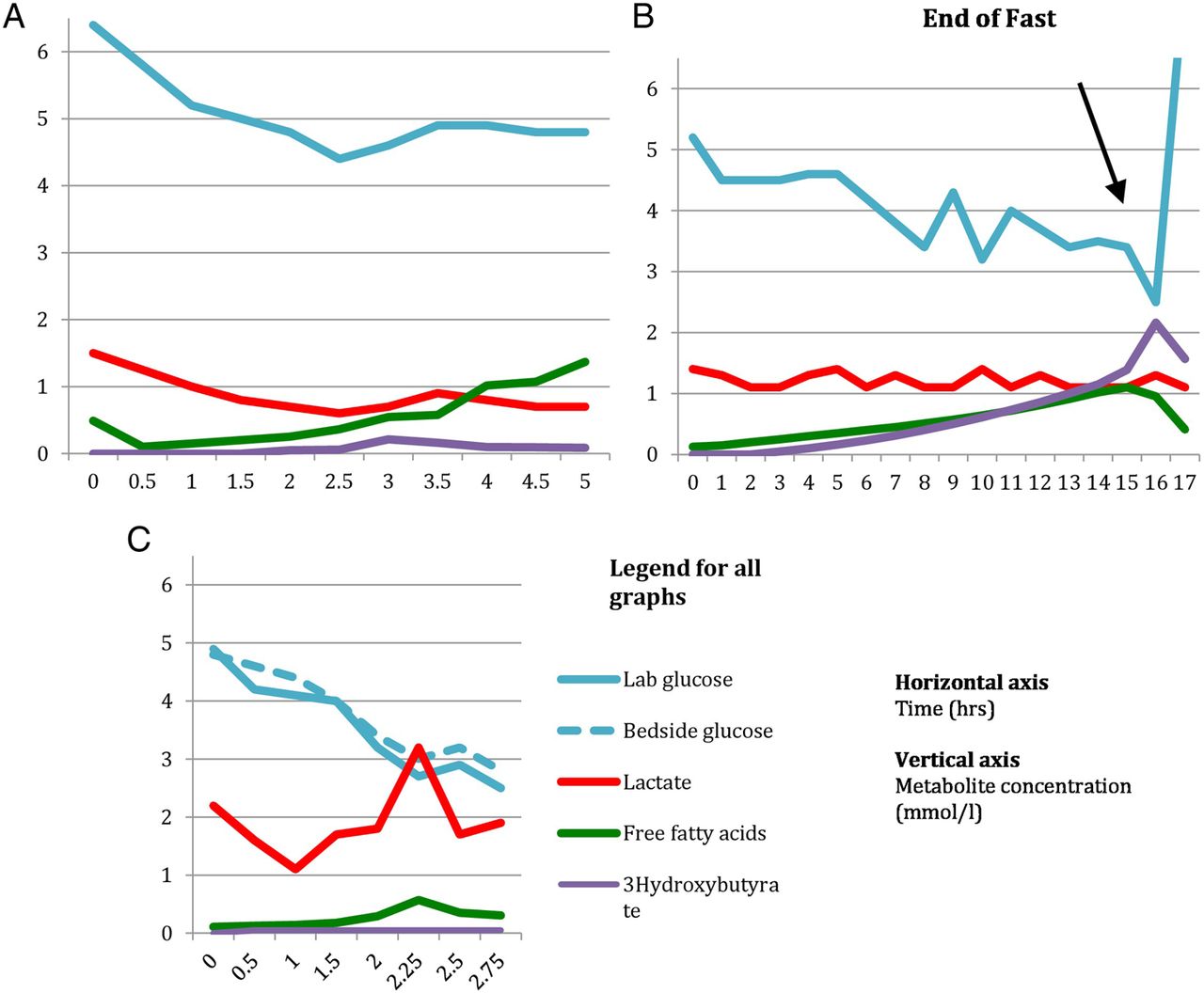
Consequently, a disturbance of any component of these responses has the potential to lead to hypoglycaemia. For instance in hyperinsulinism, high levels of insulin inhibit mobilisation of free fatty acids from adipose tissue and subsequent β-oxidation to ketones. These children typically have low levels of free fatty acids and ketone bodies at the time of hypoglycaemia. Defective β-oxidation of fatty acids in fatty acid oxidation defects explains the high levels of free fatty acids and hypoketotic hypoglycaemia noted in these children. In hepatic glycogen storage disorders, inability to mobilise glucose from liver glycogen explains the hypoglycaemia and very short fasting tolerance seen in these patients (figure 2A,C).
{kind=link}
{kind=link}
Controlled fasts scenarios.
The duration of fasting prior to hypoglycaemia can often provide clues to an underlying metabolic or endocrine disorder as shown in table 2.
Changes in predominant fuel supply during fasting
Technological background
Prerequisites for a controlled fast
It is important to exclude fatty acid oxidation defects and hyperinsulinism before subjecting a child to a controlled fast. Fasting children with fatty acid oxidation defects and hyperinsulinism can be potentially life threatening as these subjects cannot produce ketone bodies. Blood acylcarnitine analysis should always be performed and confirmed to be normal before a controlled fast. Hyperinsulinism can be diagnosed based on high glucose requirements and detectable levels of insulin at the time of hypoglycaemia.
Suspected adrenal insufficiency should be assessed by a short Synacthen test prior to the fast.
Setting
Controlled fasts should be performed according to a strict protocol in a hospital setting under close supervision. The protocol should define the threshold for termination of the fast and initiation of appropriate treatment.
Controlled fasts should be planned in advance in conjunction with the laboratory. The protocol for the controlled fast should be individualised based on several factors including the clinical presentation, age of the patient and the reason for the fast. The duration of the fast depends on the age of the child (table 3) and the start time set such that in the event of hypogylcaemia occurring, it is expected to do so during routine working hours with optimal staff levels available, for example, a child aged 8–12 months starting fasting at midnight.
Duration of fasts according to age distribution1
Patients may therefore need admission overnight according to the planned duration of the fast. The patient should be examined to ensure fitness to undergo the fast. If there are signs of intercurrent illness, the fast is better deferred until the child is well. A secure intravenous cannula must be inserted before commencing the fast to enable easy and prompt sampling without distressing the child and also to enable dextrose to be given quickly if hypoglycaemia needs treating.
During the fast, only plain water should be allowed if the child is thirsty. Specimens should be taken regularly along with bedside glucose measurements as per an individualised protocol (table 4). The clinical condition should be monitored carefully for signs of hypoglycaemia. For children admitted the night before, hourly sampling can commence at the start of the working day, that is, 08:00–16:00.
Example of specimens taken during a controlled fast
When to stop the fast?
If the patient is hypoglycaemic or symptomatic, an urgent lab glucose along with the end of fast samples should be collected and the fast terminated. In our practice, we terminate controlled fasts in an asymptomatic child if the glucometer reading and/or the true laboratory glucose reading is ≤3.0 mM.
If the patient is symptomatic, intravenous glucose should be given after the collection of end of fast samples as a slow bolus in a dose of 200 mg/kg (2 mL/kg of 10% dextrose), followed by a continuous infusion of glucose at 5–8 mg/kg/min until the patient is stable enough to tolerate oral feeds.
If the patient is asymptomatic, a meal or a high carbohydrate drink can be given.
Post end of fast samples should be taken 1–1.5 hours later.
Why collect postprandial samples?
Collection of postprandial samples is important as it can provide useful diagnostic information.
Postprandial hyperglycaemia and fasting hypoglycaemia are seen in Fanconi-Bickel syndrome, characterised by mutations within GLUT2, the gene encoding the facilitative glucose transporter in hepatocytes, pancreatic β-cells, enterocytes and renal tubular cells. Analysis of postprandial glucose, lactate and ketones can be helpful in diagnosing rare metabolic conditions like glycogen synthase deficiency (GSD type 0), where postprandial lactate and glucose are usually elevated and ketone body utilisation defects like succinyl-CoA oxoacid transferase deficiency, where ketones are not always suppressed after feeding. Abnormalities in the postprandial metabolites warrant further metabolic input and investigations.
Results from a controlled fast in a 9-month-old with glycogen storage disease type 1
Results from a controlled fast in a 3-year-old with ketotic hypoglycaemia
EXAMPLES OF CONTROLLED FAST INTERPRETATION
1) Glycogen storage disease type 1
A baby boy A aged 9 months with suspected glycogen storage disorder was electively admitted for a controlled fast to determine his safe fasting tolerance.
The results of this are shown in table 5 and figure 2C. Looking at the results of the fast, it is clear that baby A had a low plasma glucose of 2.7 mmol/L at 2 hours 15 min from the commencement of the fast. The results also illustrate how important is it to get a true glucose sample and not to rely completely on bedside glucometer readings. Had the true glucose not been checked, this child may have been assumed to be safe to fast for 30 min longer than he actually is. His safe fasting tolerance is only 2 hours.
There is no appreciable ketotic response which is also frequently seen in GSD type 1.
2) Ketotic hypoglycaemia
A boy aged 3 years with recurrent episodes of hypoglycaemia secondary to illness was admitted for a controlled fast the results of which are shown in table 6 and figure 2B.
Acylcarnitines, urine organic acids and plasma quantitative amino acids were unremarkable.
Interpretation: The results of the fast indicate that the subject was hypoglycaemic at 16 hours after the commencement of the fast. At the time of hypoglycaemia, there is an appropriate increase in the levels of free fatty acids and ketone bodies indicative of normal lipolysis and β-oxidation of fatty acids during fasting. The postprandial samples show a decrease in the levels of free fatty acids and ketone bodies, which again is in keeping with the normal physiological response.
The results also demonstrate a normal response in the levels of insulin, C peptide and growth hormone at the time of hypoglycaemia.
We can now conclude that the subject has idiopathic recurrent ketotic hypoglycaemia of childhood having excluded an underlying metabolic or endocrine cause for hypoglycaemia.
3) Fatty acid oxidation disorder
A girl aged 7 years with very long chain acyl-CoA dehydrogenase deficiency was electively admitted for a 6-hour controlled fast for assessment of her safe fasting tolerance.
The results of the fast are shown in table 7 and figure 2A.
Interpretation: Though this subject did not go hypoglycaemic during the fast, the free fatty acids show a significant rise from 4 hours onwards without corresponding ketone production. The normal molar ratio of free fatty acids to 3-hydroxybutyrate is <2. It is clear that the ratio is very high in this subject (15.7 at 5 hours).
The free fatty acids (in this patient, likely to be all long chain fatty acids) can form toxic intermediates, which can cause potentially life-threatening encephalopathy, liver failure and arrhythmias.
Therefore, the safe fasting tolerance is only 4 hours in this subject.
Results of a controlled fast in a 7 year old with VLCADD deficiency
Clinical questions
Should a controlled fast be performed in any child with suspected hypoglycaemia without a documented hyposcreen?
In children with documented or suspected hypoglycaemia, ideally the aetiology of hypoglycaemia is best investigated by analysing hormone and metabolite concentrations in blood and organic acids in urine at the time of hypoglycaemia. While many metabolic disorders can still be diagnosed from random samples, diagnosis of some, especially endocrine disorders, becomes difficult if the required samples are not taken at the time of hypoglycaemia. Under such circumstances, a controlled fast may help to elucidate the underlying cause of hypoglycaemia. If there is no clear evidence of a metabolic or endocrine defect from the baseline tests, the decision to do a controlled fast should be based on the age of the child, presence of symptoms and the frequency of attacks.
An acylcarnitine profile should always be analysed prior to organising a controlled fast as fasting can be dangerous in children with fatty acid oxidation defects. Impaired ketogenesis in these children results in accumulation of fatty acids and toxic acylcarnitine metabolites, which can lead to encephalopathy and death if not treated promptly. There are case reports of children with medium chain acyl-CoA dehydrogenase deficiency, who have died as a result of prolonged controlled fasts and those with very long chain acyl-CoA dehydrogenase deficiency, where fasting provoked rhabdomyolysis.1 Fatty acid oxidation defects can now be diagnosed with the help of tandem mass spectrometry which is a much safer method.
Morris et al1 reviewed 138 controlled fasts organised over a 30-month period in a tertiary metabolic centre. 54 (39%) resulted in hypoglycaemia and a specific endocrine or metabolic cause was identified in 30 (21% of all fasts) patients. The diagnostic yield was highest in young children, specific defects being identified in 14 of the 32 children fasted at the age of <1 year. As expected, disorders were identified more frequently if hypoglycaemia had been documented at the referring hospital than if it was merely suspected. The diagnostic yield now would be expected to be lower due to the wider availability of metabolic tests such as acylcarnitine profiles that can diagnose inborn errors of metabolism without the need for controlled fasting.
Controlled fasts can also help the family and clinician in determining the safe fasting tolerance of the child when well and tailor dietary management.
Clinical bottom line
Diagnosis of endocrine disorders causing hypoglycaemia may be missed if samples are not taken at the time of hypoglycaemia.
In the absence of a hypoglycaemia screen, a controlled fast should be undertaken based on the individual clinical situation. It can be valuable in determining the maximum safe interval between feeds and prevent further hypoglycaemic episodes.
Acylcarnitine profile should be confirmed to be normal prior to organising a controlled fast.
Is a controlled fast indicated in a neonate with high glucose requirement and in whom the opportunity was missed to do a hypoglycaemia screen?
The normal glucose requirement in a neonate is 4–8 mg/kg/min. Hyperinsulinism should be considered in a neonate when the glucose requirement is >10 mg/kg/min. It should be considered in any neonate with persistent or frequent hypoglycaemic episodes.
Hyperinsulinism is a predominant cause of profound hypoglycaemia in the neonatal period, which, if untreated, can lead to significant morbidity and mortality. Studies have shown that recurrent spontaneous hypoglycaemia may not be accompanied by recognised neuroglycopenic symptoms in infants with congenital hyperinsulinism.5 Therefore, it is important to have a high index of suspicion of hyperinsulinism in a neonate with hypoglycaemia and high glucose requirement.
Hyperinsulinism is diagnosed by the finding of detectable insulin levels, low free fatty acid concentrations and low ketones at the time of hypoglycaemia. Hence, a hypoglycaemia screen would be ideal in diagnosing hyperinsulinism.
A controlled fast is not necessarily indicated in neonates with hyperinsulinism in whom the diagnosis can be made based on the high glucose requirement and widely available assays to measure insulin at the time of hypoglycaemia. Fasting can be dangerous in these subjects as they cannot produce ketones, which can be used as alternative sources of energy supply. Specialist endocrine input should be sought in all cases of suspected hyperinsulinism, who did not have a hyposcreen, to guide further management.
Clinical bottom line
Hyperinsulinism should be considered in any neonate with persistent or frequent hypoglycaemic episodes and in whom the glucose requirement is >10 mg/kg/min.
Close liaison with an endocrinologist is useful in managing hypoglycaemia secondary to hyperinsulinism, especially the refractory cases who may need specific therapy as diazoxide and chlorthiazide.
Is a controlled fast indicated in a child with hypoglycaemia secondary to illness?
Hypoglycaemia secondary to an intercurrent illness like diarrhoea and vomiting in a child is a common scenario in paediatric emergency department. Ideally, every attempt should be made to collect crisis samples at the time of hypoglycaemia and the next available urine collected for analysis of ketones and organic acids.
However, it is important to establish whether children are ketotic at the time of hypoglycaemia. In the review by Morris et al,1 32 (59%) children who became hypoglycaemic during the fast had ‘ketotic hypoglycaemia’.
Idiopathic ketotic hypoglycaemia (IKH) is the most frequent term used to describe recurrent hypoglycaemia in children aged between 1 and 5 years.13 The clinical pattern was characterised by Colle and Ulstrom as the tendency of affected children to become ketotic and hypoglycaemic during a low calorie ketogenic diet.11 It is due to the failure of hepatic glucose production to meet the body's increase in glucose demand during illness or fasting.13 ,14 IKH typically improves with age and is not usually observed after children reach puberty.
Recurrent episodes necessitate baseline metabolic investigations including an acylcarnitine profile prior to controlled fast. In cases with recurrent episodes or symptomatic hypoglycaemia, metabolic specialist opinion should be sought regarding further management. X-linked adrenoleukodystrophy should always be considered in a boy with recurrent episodes of hypoglycaemia and low cortisol levels.
Controlled fasts should be considered in children with clinical features of a primary endocrine or metabolic defect or in those with recurrent symptomatic hypoglycaemia.
Controlled fasts can also be helpful in determining the fasting tolerance in suspected cases of primary endocrine or metabolic disorder.
Clinical bottom line
IKH is common in childhood but it is a diagnosis of exclusion.
In the absence of a hyposcreen, the decision to undertake a controlled fast should be taken based on individual clinical situation and after close liaison with metabolic specialists.
Summary
Controlled fasts can play a valuable role in the diagnosis and management of hypoglycaemia in paediatric clinical practice, but are no substitute for the collecting of appropriate critical samples at the time of hypoglycaemia for metabolic and endocrine studies. Fatty acid oxidation defects, hyperinsulinism and adrenal insufficiency should always be excluded prior to organising controlled fasts.
Controlled fasts are safe if conducted in an experienced setting with strict protocols in place. Failure to adhere to protocol can defeat the purpose of the study and can potentially be dangerous.
Proper planning in conjunction with the laboratory and close supervision by staff experienced in controlled fasts is crucial to ensure the best quality information is yielded from these procedures.
References
Footnotes
Contributors SSr: written the article. SSa: contributed to design and presentation of the data, gave ideas for the scenarios, critically reviewed the paper and finalised it. JR: reviewed the clinical questions and also helped with the design and presentation of the data. SV: reviewed the paper. MAP: reviewed relevant literature, contributed to design and presentation of the data and offered ideas for the scenario.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.