Article Text

Abstract
Tuberous sclerosis complex is a complex childhood disorder characterised by the formation of hamartomas in multiple organs. Annual review of this disease is recommended to monitor the development of complications. We aim to provide a concise, evidence-based framework to assist clinicians during this annual review. We focus on the following areas: (A) what questions need to be asked during annual review, (B) which areas need emphasis on examination, (C) when is an investigation required and (D) referral to tertiary specialists and other members of the multidisciplinary team. It should also be noted that there are ongoing debates regarding screening intervals in certain areas. These include the frequency, modality of screening and degree of intervention for astrocytomas and renal angiomyolipomas. This review seeks to summarise the product of the ongoing debates, and provide evidence-based suggestions in light of the uncertainty.
- Paediatric Practice
- Neurology
- Dermatology
- Nephrology
- Adolescent Health
Statistics from Altmetric.com
Aim
Tuberous sclerosis complex (TSC) is a genetically determined disorder characterised by the formation of hamartomas in multiple organs, affecting 1:5000–1:10 000 live births. The most common organs involved are the brain, skin, kidneys, retina and heart.1–5
Annual surveillance of children with tuberous sclerosis is recommended to monitor the multiorgan manifestations. This can be daunting to paediatricians due to the complexity and uncertainty of the natural progression of the disease. This review does not cover all aspects of TSC. However, we recognise the importance of an overview of the pathogenesis and clinical manifestations, and we shall cover them briefly.
We aim to provide a concise, evidence-based framework to assist secondary level, community and acute paediatricians during a 30–60 min consultation. We focus on the following areas: (A) what questions need to be asked during annual review, (B) which areas need emphasis on examination, (C) when is an investigation required and (D) referral to tertiary specialists and other members of the multidisciplinary team. Ongoing debates regarding screening intervals remain in certain areas. These include the frequency, modality of screening and degree of intervention for astrocytomas and renal angiomyolipomas (AML). This review seeks to summarise the product of the ongoing debates, and provide evidence-based suggestions in light of the uncertainty.
While this article is targeted at the review of children with TSC, there is heterogeneity in presentation and different issues to be addressed across a broad age range. We have provided a timeline to highlight the natural progression of the disease, providing a guide to the paediatrician on which areas need emphasis across a range of ages (figure 1).
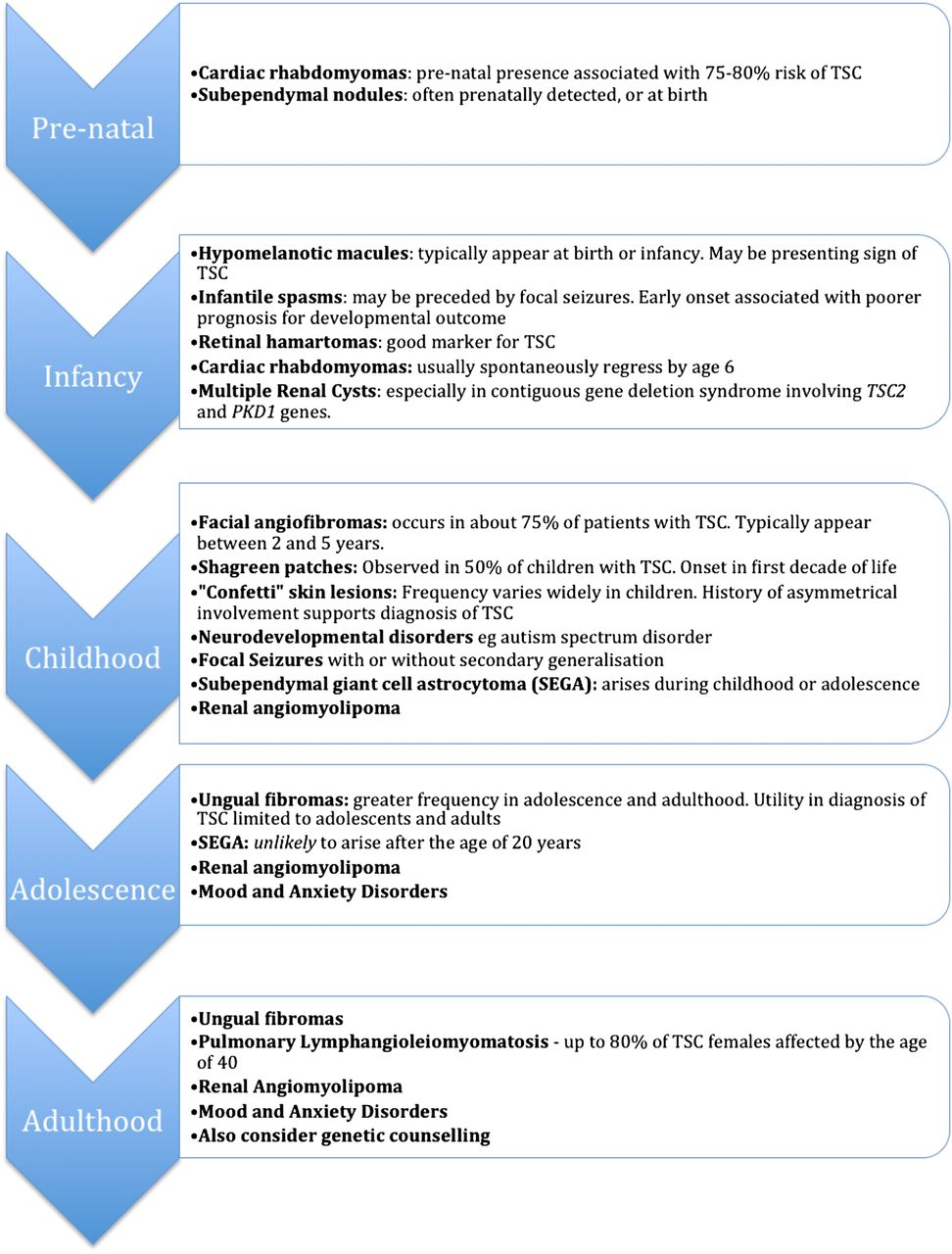
Timeline of various manifestations of TSC.6 TSC, tuberous sclerosis complex; PKD, polycystic kidney disease.
Genetics of tuberous sclerosis
TSC exhibits autosomal-dominant inheritance. Two distinct loci have been identified: chromosomes 9q34 (TSC1) and 16p13 (TSC2), facilitating the production of hamartin and tuberin, respectively. Together, they form a heterodimer which suppresses the mammalian target of rapamycin (mTOR), which is involved in increasing cell proliferation and growth. Hence, inactivating mutations on TSC loci cause dysregulation of cell proliferation.1
Confirming the diagnosis
TSC is diagnosed by a range of clinical criteria, as displayed in table 1.
Diagnostic criteria for tuberous sclerosis complex (TSC)6
Clinicians should consider genetic screening for pathogenic mutations in suspicious cases that do not fulfil the required clinical criteria. In clinically confirmed cases, genetic screening may be useful if counselling is required for future family planning. It also offers prognostic information as mutations in TSC2 tend to result in more severe phenotypes.
Neurological manifestations
Epileptic seizures
The most common presentation is with epilepsy occurring during the first year of life. The two main types of TSC-associated epilepsy are focal seizures and infantile spasms. Most children with infantile spasms develop at least another seizure phenotype or even multiple seizure phenotypes. Early onset of epilepsy (especially infantile spasms) is associated with cognitive and behavioural impairments.2 All affected children should be referred to an epilepsy specialist.
A baseline electroencephalogram (EEG) should be performed in all children diagnosed with TSC regardless of whether clinical seizures have been reported. Care should be taken to treat the child and their seizures, rather than the EEG in isolation.7 A low threshold for a repeat should be maintained, especially with an unexplained decline of behavioural or cognitive function.
We recommend the following during annual surveillance for children with TSC not known to have seizures:
-
Has there been any evidence of tonic/clonic, absence or other types of seizures in the past year?
-
If the answer to any of the above is ‘yes’, we would recommend referral to an epilepsy specialist with the consideration for a repeat EEG and 24 h video EEG.
-
Parents should be educated to recognise seizure types irrespective of seizure history.
For children with TSC diagnosed with epilepsy or suspected of seizure activity, EEG frequency is dependent on clinical need (frequency/severity of seizures). The paediatrician should review current antiepileptic medication. Annual screening for tuberous sclerosis associated neuropsychiatric disorders (TAND) should be performed, and any unexplained changes should be followed up with prolonged video EEG.5 ,6
The choice of treatment is age-related and differs depending on the type of seizure; for instance, infantile spasms in patients with TSC are normally treated with vigabatrin initially despite concerns of visual field constriction.8 Risks of complications should be minimised by initiating low doses of the drug for brief periods.
Neurological tubers
Brain lesions in TSC include tubers in the cerebral cortex and subependymal nodules (SEN) along the walls of the lateral ventricles. The number of cortical tubers partially correlates with learning disability and epilepsy, but are not associated with oncological growth potential. SENs usually remain dormant, but may increase in size, developing into a subependymal giant cell astrocytoma (SEGA) which can cause hydrocephalus. SEGAs occur in about 6%–14% of patients with TSC, can present within the first years, but rarely start to grow after the age of 20 years.
Factors increasing risk of a SEN transforming into SEGA include9:
-
location: near the foramen of Monro;
-
appearance: incompletely calcified and enhanced by gadolinium;
-
size: >5 mm.
O'Callaghan et al10 demonstrated the incidence of symptomatic SEGA in patients with TSC to be low but significant—5.6% in a population-based study. Key parts of the annual check should aim to identify the development of symptoms related to ventricular enlargement or TAND symptoms:
-
History: enquire about symptoms of raised intracranial pressure (ICP) and signs that increase suspicion of SEGAs—headaches, vomiting, head banging, increased frequency of seizures, behavioural changes or visual changes.
-
Examination: look for focal neurological signs, funduscopy for papilloedema and other signs of raised ICP.
-
If raised ICP is suspected, an urgent MRI is required. A high index of suspicion should be maintained for patients with learning difficulties.
We would recommend an MRI with contrast at the point of diagnosis.
Subsequent screening for SEGAs remains one of the most debated points in surveillance of children with TSC. A summary of the evidence and arguments is shown in table 2. O'Callaghan et al10 proposed that regular screening was not indicated as intervention is normally not required without symptoms, noting that assessment of headache could be challenging in children with learning disabilities. More recently, the 2013 statement from the TSC Consensus Meeting for SEGA and Epilepsy Management recommends two yearly scans throughout childhood.11 On balance, we recommend a 2-yearly MRI head with gadolinium throughout childhood. Patients with large or growing SEGAs, who remain asymptomatic should undergo more frequent MRI scans. All the images, and situations where it is difficult to perform the scan (eg, older children with severe autism), should be discussed with a tertiary specialist, who will counsel the family on the risks versus benefits of less frequent scans.
Neurodevelopmental, psychiatric and cognitive problems
Approximately 55% of children with TSC have cognitive function within the normal range, but 30% have global intellectual ability in the severe to profoundly impaired range. Developmental outcome and progress is correlated with age of onset, severity and control of seizure in some children. A high index of suspicion needs to be maintained for all children who develop TAND symptoms (table 3) at a young age, especially under the age of 3 years.5 While learning disability is a classic and common feature of TSC, autism, attentional deficits and other difficulties may be missed.
TSC-associated neuropsychiatric disorders (TAND)
Psychiatric and behavioural disturbances
De Vries et al13 recommended that pervasive developmental disorders be investigated at 2 years and again at school entry age of 4–5 years. Multiple tools can be used for a more objective evaluation, including the Childhood Autism Test (CHAT) and Alzheimer's Disease Information Questionnaire (ADIQ) tests.
Renal manifestations
Eighty per cent of children with TSC develop renal abnormalities, including renal cystic disease, AML, polycystic kidney disease (PKD) and, extremely rarely in childhood, renal cell carcinoma. The PKD1 gene is contiguous with TSC2, so only children with contiguous gene deletions around TSC2 are at increased risk of PKD. TSC abnormalities increase in incidence and number with age. While the majority of these lesions are asymptomatic before adolescence, renal involvement can lead to hypertension and chronic kidney disease in severe, untreated cases. The formation of aneurysms in renal AML also exposes patients to the risk of life-threatening haemorrhage.4 The mean age for development of renal abnormalities is between 7.2 and 9.2 years.14
Most consensus panels accept that abdominal imaging (ultrasound, but MRI also supported5) should be performed at diagnosis to evaluate the presence of renal lesions. If normal, we recommend the following for annual review (figure 2):
-
History: symptoms associated with bleeding from AML—back or abdominal pain, haematuria, nausea, vomiting, fever, irritability.
-
Examination: renal enlargement, blood pressure.
-
Investigations: if asymptomatic, we recommend a yearly or 2-yearly renal ultrasound, or an MRI (better delineation), from the age of 8 years. This is because it is unlikely for AMLs identified under the age of 8 years to require any intervention due to their early stage.

Flowchart for surveillance of renal manifestations in TSC. TSC, tuberous sclerosis complex; US, ultrasound.
All abnormal scans should be reviewed by a TS specialist. In practice, most experts will only intervene if the size exceeds 3.5 cm.15 Therefore, where the AML is under 3.5 cm wide and asymptomatic, it is likely no further action will be taken, other than reviewing scan frequency and earlier assessment, if symptomatic. Unless the patient is on an mTOR inhibitor, patients with hypertension should be treated with an inhibitor of the renin–aldosterone–angiotensin system.
Cardiac manifestations
Cardiac rhabdomyomas are common in neonates but rarely cause complications. They occur in around 60% of children with TS, but only 20% of adults. This is attributed to spontaneous tumour regression, particularly in the first 6 years of life.16
An echocardiogram and ECG should be performed at diagnosis to detect cardiac rhabdomyomas and arrhythmias. During annual review, the paediatrician should enquire about symptoms, such as chest pain, palpitations, dyspnoea and loss of consciousness, and perform a cardiovascular examination.
Most cardiac rhabdomyomas are asymptomatic, with a minority experiencing arrhythmias (usually Wolff–Parkinson–White syndrome) and/or cardiac failure. If a rhabdomyoma is present, follow-up echocardiograms should be performed one to three-yearly in asymptomatic patients until regression is documented. A 12-lead ECG every 3–5 years is also recommended. These are performed more frequently in children with symptoms, abnormal echocardiogram/ECG, or additional risk factors such as patients treated with adrenocorticotropic hormone.5
Treatment is only indicated when the tumour causes symptomatic outflow obstruction and/or arrhythmia. Surgical removal is technically difficult and controversial, because rhabdomyomas are located deep in the myocardium.17 An urgent referral to a paediatric cardiologist should be made if symptoms or signs are present, or if the patient is known to have multiple high-risk rhabdomyomas at diagnosis.
Pulmonary manifestations
Lymphangioleiomyomatosis (LAM) is a slowly progressive cystic lung disease associated with mutations in the TSC2 gene. The average age at diagnosis of LAM is 35 years, with the risk increasing by ∼8% per year.18 Studies have found evidence of pulmonary involvement in up to 35% of women with TSC.
LAM is very rare among the under-16 s, and there is currently no recommendation for routine testing in asymptomatic children. In view of its age-dependent risk, asymptomatic women above the age of 18 years should receive a high-resolution CT (HRCT) every 5–10 years if there is no evidence of lung cysts at baseline imaging.5 Individuals with lung cysts are recommended to undergo annual pulmonary function tests as well as more frequent HRCT. Counselling regarding risks from smoking and oestrogen use is also recommended.
Dermatological manifestations
The most common skin changes seen in TSC are hypomelanotic macules, angiofibromas, shagreen patches, forehead plaques and ungual fibromas, as displayed in table 4.19 Recognition of the key lesions is important, as they can appear anytime during childhood and can cause anxiety.
Skin manifestations in tuberous sclerosis*
During annual review, clinicians should enquire about progression of known skin lesions and the development of new ones, particularly the effects on the child's quality of life. Skin inspection should be performed. Reassurance and a comforting discussion are often sufficient. However, if these lesions are debilitating (eg, cosmetically disfiguring or bleeding), referral to the dermatologist is indicated.
Ophthalmological manifestations
Approximately half the children with TSC have retinal astrocytic (RA) hamartomas. Two studies showed that there was no correlation between age and type of retinal hamartomas, supporting the premise that most retinal lesions in TSC remain stable over time.20
Children with known TSC are at risk of ophthalmic complications such as squint, and should have community vision screening between the ages of 4 and 4.5 years. Early referral to the hospital eye service is warranted if a squint or reduced vision is suspected.
For the purposes of annual review, RA hamartomas are congenital lesions that do not increase in size and number. Thus, regular retinal examination is not required unless ophthalmic issues such as squint, coloboma or papilloedema are present.
Transition
Active planning needs to be undertaken from the age of 14 years, for transition through to adult services when the individual reaches the age of 16. The young adult, family and general practitioner should be empowered to take ownership of subsequent surveillance, with referral to specialist services, such as those for epilepsy and adult learning disability, only where needed. The paediatrician, with help from the tertiary TS specialist, should summarise the recommendations in a format allowing understanding by the young adult (depending on their mental age), and appropriate carers.
Concluding the consultation
The family should be given the floor at the end of the consultation to ask questions. TSC, with its multiple complex manifestations, can cause substantial parental anxiety and can prove to be a social, emotional and economic burden to the family. The role of patient organisations such as the TS Association can be invaluable. Alongside the welfare concerns is the ongoing research with mTOR inhibitors such as everolimus, which at its initial stages, has proven helpful in shrinking SEGAs, AML and rhabdomyomas.
We have provided an illustrated summary of our suggestions for annual review in Figure 3. We hope that this article can provide some answers to questions from parents; but very often their questions will be beyond its scope. The paediatrician should have a low threshold to ‘phone a friend’, especially the regional TS expert.
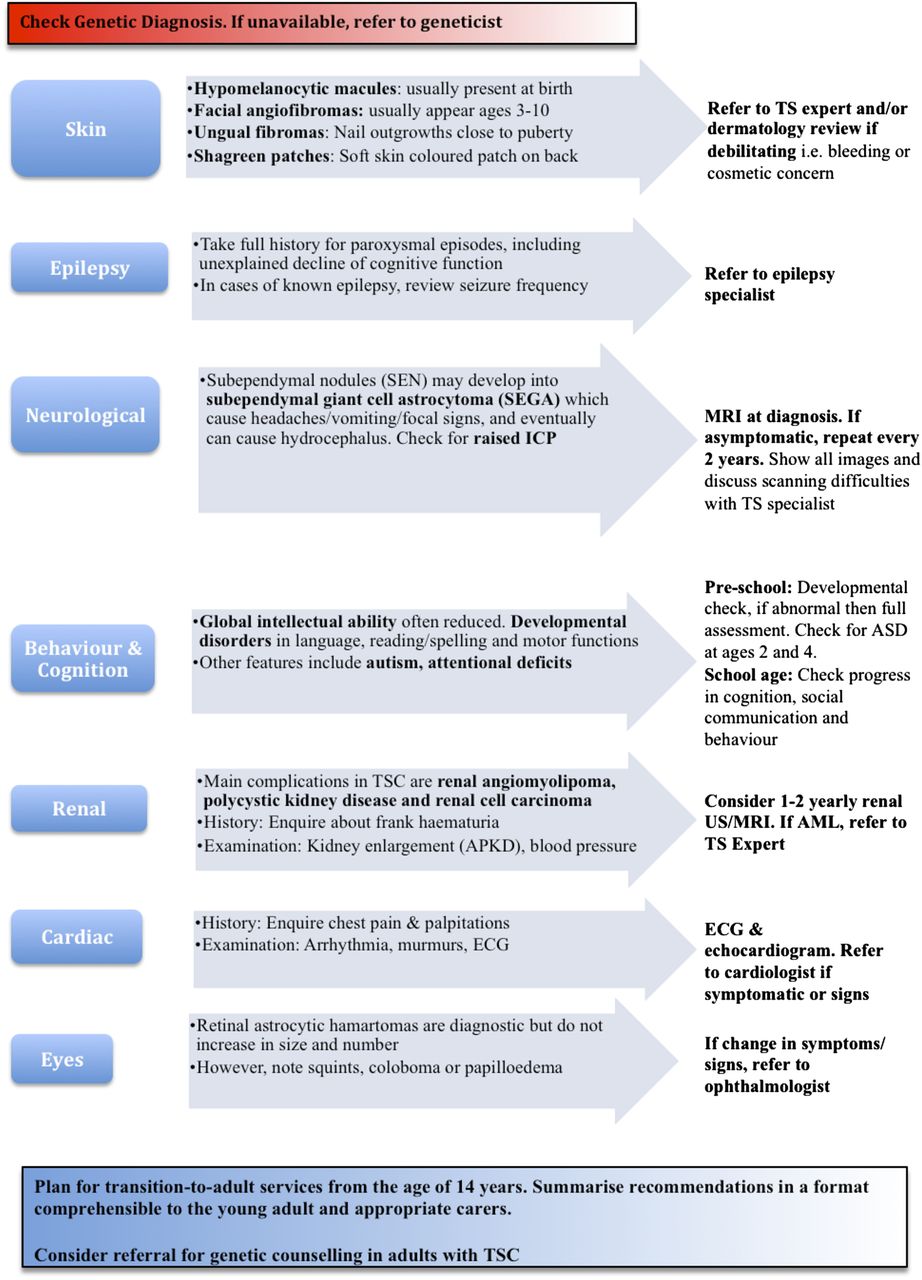
{kind=link}
{kind=link}
{kind=link}
Annual review of checklist for children with TSC. (A complete checklist and a more detailed version of the evidence used to produce this review are available online via the Eastern Paediatric Epilepsy Network: http://www.networks.nhs.uk/nhs-networks/eastern-paediatric-epilepsy-network. We also hope that paediatricians will find the updated proforma in the website useful during review of patients with tuberous sclerosis). TSC, tuberous sclerosis complex; ICP, intracranial pressure; US, ultrasound; AML, angiomyolipoma; ASD, autism spectrum disorder.
References
Footnotes
-
Contributors The three authors are justifiably credited with authorship according to the ICMJE authorship criteria. In detail, KHN: conception, design, analysis and interpretation of data, drafting of manuscript, providing final approval; SMN: acquisition of data, conception and design of work, drafting of manuscript, providing final approval; AP: conception, design, analysis and interpretation of data, drafting of the manuscript, critical revision of manuscript, providing final approval.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.